ANNUAL
INFORMATION FORM
Fiscal
year ended May 31, 2007
August
29, 2007
2
Meridian Road, Toronto, Ontario M9W 4Z7
Telephone: (416)
798-1200
Fax:
(416) 798-2200
TABLE
OF CONTENTS
|
CAUTION
REGARDING FORWARD-LOOKING STATEMENTS
|
1
|
|||
|
THE
COMPANY
|
4
|
|||
|
GENERAL
DEVELOPMENT OF THE BUSINESS
|
4
|
|||
|
REGULATORY
REQUIREMENTS
|
7
|
|||
|
BUSINESS
OF THE COMPANY
|
10
|
|||
|
Overview
|
10
|
|||
|
Antisense-DNA/RNA-based
Therapeutics
|
10
|
|||
|
Small
Molecule Therapies
|
15
|
|||
|
Immunotherapy
|
16
|
|||
|
Other
Technologies
|
18
|
|||
|
Agreements
|
19
|
|||
|
Business
Strategy
|
21
|
|||
|
Financial
Strategy
|
22
|
|||
|
Intellectual
Property and Protection of Confidential Information and
Technology
|
24
|
|||
|
Regulatory
Strategy
|
25
|
|||
|
Competition
|
25
|
|||
|
Human
Resources
|
26
|
|||
|
Properties
|
26
|
|||
|
Control
of the Registrant
|
26
|
|||
|
RISK
FACTORS
|
26
|
|||
|
RISKS
RELATED TO OUR COMMON SHARES AND CONVERTIBLE DEBENTURES
|
26
|
|||
|
DIVIDENDS
|
34
|
|||
|
SHARE
CAPITAL AND MARKET FOR SECURITIES
|
35
|
|||
|
Share
Capital
|
35
|
|||
|
Market
for Securities
|
35
|
|||
|
Principal
Shareholders
|
35
|
|||
|
DIRECTORS
AND OFFICERS
|
36
|
|||
|
COMMITTEE
INFORMATION
|
37
|
|||
|
Audit
Committee
|
37
|
|||
|
Independent
Auditors
|
37
|
|||
|
LEGAL
PROCEEDINGS
|
38
|
i
|
TRANSFER
AGENT AND REGISTRAR
|
38
|
|||
|
MATERIAL
CONTRACTS
|
39
|
|||
|
INTERESTS
OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
|
40
|
|||
|
INTEREST
OF EXPERTS
|
40
|
|||
|
ADDITIONAL
INFORMATION
|
40
|
|||
|
GLOSSARY
|
42
|
|||
|
CHARTER
OF THE AUDIT COMMITTEE OF THE BOARD OF DIRECTORS OF LORUS THERAPEUTICS
INC. (THE “COMPANY”)
|
46
|
ii
CAUTION
REGARDING FORWARD-LOOKING STATEMENTS
This
annual information form may contain forward-looking statements within the
meaning of Canadian and U.S. securities laws. Such statements
include, but are not limited to, statements relating to:
|
|
•
|
our
expectations regarding future
financings;
|
|
|
•
|
our
plans to conduct clinical
trials;
|
|
|
•
|
our
expectations regarding the progress and the successful and timely
completion of the various stages of our drug discovery, preclinical
and
clinical studies and the regulatory approval
process;
|
|
|
•
|
our
plans to obtain partners to assist in the further development of
our
product candidates; and
|
|
|
•
|
our
expectations with respect to existing and future corporate alliances
and
licensing transactions with third parties, and the receipt and
timing of
any payments to be made by us or to us in respect of such arrangements,
and
|
the
Company’s plans, objectives, expectations and intentions and other statements
including words such as “anticipate”, “contemplate”, “continue”, “believe”,
“plan”, “estimate”, “expect”, “intend”, “will”, “should”, “may”, and other
similar expressions.
Such
statements reflect our current views with respect to future events and are
subject to risks and uncertainties and are necessarily based upon a number
of
estimates and assumptions that, while considered reasonable by us are inherently
subject to significant business, economic, competitive, political and social
uncertainties and contingencies. Many factors could cause our actual results,
performance or achievements to be materially different from any future results,
performance, or achievements that may be expressed or implied by such
forward-looking statements, including, among others:
|
|
•
|
our
ability to obtain the substantial capital required to fund research
and
operations;
|
|
|
•
|
our
lack of product revenues and history of operating
losses;
|
|
|
•
|
our
early stage of development, particularly the inherent risks and
uncertainties associated with (i) developing new drug candidates
generally, (ii) demonstrating the safety and efficacy of these
drug
candidates in clinical studies in humans, and (iii) obtaining regulatory
approval to commercialize these drug
candidates;
|
|
|
•
|
our
drug candidates require time-consuming and costly preclinical and
clinical
testing and regulatory approvals before
commercialization;
|
|
|
•
|
clinical
studies and regulatory approvals of our drug candidates are subject
to
delays, and may not be completed or granted on expected timetables,
if at
all, and such delays may increase our costs and could delay our
ability to
generate revenue;
|
|
|
•
|
the
regulatory approval process;
|
|
|
•
|
the
progress of our clinical
trials;
|
|
|
•
|
our
ability to find and enter into agreements with potential
partners;
|
|
|
•
|
our
ability to attract and retain key
personnel;
|
|
|
•
|
our
ability to obtain patent protection and protect our intellectual
property
rights;
|
|
|
•
|
our
ability to protect our intellectual property rights and to not
infringe on
the intellectual property rights of
others;
|
|
|
•
|
our
ability to comply with applicable governmental regulations and
standards;
|
|
|
•
|
development
or commercialization of similar products by our competitors, many
of which
are more established and have greater financial resources than
we
do;
|
|
|
•
|
commercialization
limitations imposed by intellectual property rights owned or controlled
by
third parties;
|
|
|
•
|
our
business is subject to potential product liability and other
claims;
|
|
|
•
|
our
ability to maintain adequate insurance at acceptable
costs;
|
|
|
•
|
further
equity financing may substantially dilute the interests of our
shareholders;
|
|
|
•
|
changing
market conditions; and
|
|
|
•
|
other
risks detailed from time-to-time in our ongoing quarterly filings,
annual
information forms, annual reports and annual filings with Canadian
securities regulators and the United States Securities and Exchange
Commission, and those which are discussed under the heading “Risk
Factors”.
|
Should
one or more of these risks or uncertainties materialize, or should the
assumptions set out in the section entitled “Risk Factors” underlying those
forward-looking statements prove incorrect, actual results may vary materially
from those described herein. These forward-looking statements are made as
of the
date of this annual information form or, in the case of documents incorporated
by reference herein, as of the date of such documents, and we do not intend,
and
do not assume any obligation, to update these forward-looking statements,
except
as required by law. We cannot assure you that such statements will prove
to be
accurate as actual results and future events could differ materially from
those
anticipated in such statements. Investors are cautioned that forward-looking
statements are not guarantees of future performance and accordingly investors
are cautioned not to put undue reliance on forward-looking statements due
to the
inherent uncertainty therein.
Unless
otherwise indicated, or the context requires otherwise, the information
appearing in this annual information form is stated as at May 31, 2007 and
references in this annual information form to “$” or “dollars” are to Canadian
dollars.
On
July 10, 2007 (the “Arrangement Date”), the Old Lorus completed a plan of
arrangement and corporate reorganization with, among others, 6650309 Canada
Inc.
(“New Lorus”), 6707157 Canada Inc. and Pinnacle International Lands,
Inc. As a result of the plan of arrangement and reorganization, among
other things, each common share of Old Lorus was exchanged for one common
share
of New Lorus and the assets (excluding certain future tax attributes and
related
valuation allowance) and liabilities of Old Lorus (including all of
the
-
2
-
shares
of its subsidiaries held by it) were transferred, directly or indirectly,
to the
Company and/or its subsidiaries. New Lorus continued the business of
Old Lorus after the Arrangement Date with the same officers and employees
and
continued to be governed by the same board of directors as Old Lorus prior
to
the Arrangement Date. References in this annual information form to the Company,
Lorus, “we”, “our”, “us” and similar expressions, unless otherwise stated, are
references to Old Lorus prior to the Arrangement Date and New Lorus after
the
Arrangement Date.
For
ease of reference, a glossary of terms used in this annual information form
can
be found beginning on page 42.
-
3
-
THE
COMPANY
Lorus
Therapeutics Inc. (“Old Lorus”) was incorporated under the Business
Corporations Act (Ontario) on September 5, 1986 under the name RML Medical
Laboratories Inc. On October 28, 1991, RML Medical Laboratories Inc.
amalgamated with Mint Gold Resources Ltd., resulting in Old Lorus becoming
a
reporting issuer (as defined under applicable securities law) in Ontario,
on
such date. On August 25, 1992, Old Lorus changed its name to IMUTEC
Corporation. On November 27, 1996, Old Lorus changed its name to
Imutec Pharma Inc., and on November 19, 1998, Old Lorus changed its name
to
Lorus Therapeutics Inc. On October 1, 2005, Old Lorus continued under
the Canada Business Corporations Act.
On
July 10, 2007 (the “Arrangement Date”), Old Lorus completed a plan of
arrangement and corporate reorganization with, among others, 6650309 Canada
Inc.
(“New Lorus”), 6707157 Canada Inc. and Pinnacle International Lands,
Inc. As a result of the plan of arrangement and reorganization, among
other things, each common share of Old Lorus was exchanged for one common
share
of New Lorus and the assets (excluding certain future tax attributes and
related
valuation allowance) and liabilities of Old Lorus (including all of the shares
of its subsidiaries held by it) were transferred, directly or indirectly,
to the
Company and/or its subsidiaries. New Lorus continued the business of
Old Lorus after the Arrangement Date with the same officers and employees
and
continued to be governed by the same board of directors as Old Lorus prior
to
the Arrangement Date. References in this annual information form to the Company,
Lorus, “we”, “our”, “us” and similar expressions, unless otherwise stated, are
references to Old Lorus prior to the Arrangement Date and New Lorus after
the
Arrangement Date.
The
address of the Company’s head and registered office is 2 Meridian Road, Toronto,
Ontario, Canada, M9W 4Z7. Our corporate website is
www.lorusthera.com. The contents of the website are specifically not
included in this annual information form by reference.
Our
common shares are listed on the Toronto Stock Exchange under the symbol “LOR”
and are listed on the American Stock Exchange under the symbol
“LRP”.
Lorus’
subsidiaries are GeneSense Technologies Inc. (“GeneSense”), a corporation
incorporated under the laws of Canada, of which Lorus owns 100% of the issued
and outstanding share capital, and NuChem Pharmaceuticals Inc. (“NuChem”), a
corporation incorporated under the laws of Ontario, of which Lorus owns 80%
of
the issued and outstanding voting share capital and 100% of the issued and
outstanding non-voting preference share capital.
GENERAL
DEVELOPMENT OF THE BUSINESS
Lorus
Therapeutics Inc. is a life sciences company focused on the discovery, research
and development of effective anticancer therapies with a high safety profile.
Lorus has worked to establish a diverse, marketable anticancer product pipeline,
with products in various stages of development ranging from preclinical to
multiple Phase II clinical trials. A growing intellectual property portfolio
supports our diverse product pipeline.
Our
success is dependent upon several factors, including establishing the efficacy
and safety of our products in clinical trials, securing strategic partnerships,
obtaining the necessary regulatory approvals to market our products and
maintaining sufficient levels of funding through public and/or private
financing.
We
believe that the future of cancer treatment and management lies in drugs
that
are effective, safe and have minimal side effects, and therefore improve
a
patient's quality of life. Many of the cancer drugs currently approved for
the
treatment and management of cancer are toxic with severe side effects, and
we
therefore believe that a product development plan based on effective and
safe
drugs could have broad
-
4
-
applications
in cancer treatment. Lorus' strategy is to continue the development
of our product pipeline using several therapeutic approaches. Each therapeutic
approach is dependent on different technologies, which we believe mitigates
the
development risks associated with a single technology platform. We evaluate
the
merits of each product throughout the clinical trial process and consider
commercialization as appropriate. The most advanced anticancer drugs in our
pipeline, each of which flow from different platform technologies, are
antisense-DNA/RNA-based therapeutics, small molecules and
immunotherapeutics.
Over
the past three years, we have focused on advancing our product candidates
through pre-clinical and clinical testing. You should be aware that it can
cost
millions of dollars and take many years before a product candidate may be
approved for therapeutic use in humans. In addition, a product candidate
may not
meet the end points of any Phase I, Phase II or Phase III clinical trial.
See
“Risk Factors”.
Antisense-DNA/RNA-based
Therapeutics
Our
lead antisense product in clinical development is GTI-2040. In addition we
have
a number of other antisense molecules in development. See “-- Clinical
Development” and “Business of the Company - Antisense-DNA/RNA-based
Therapeutics” for more details.
GTI-2040
Seven
of the nine clinical studies for GTI-2040 have been conducted in conjunction
with the United States National Cancer Institute (“NCI”) with the remaining
studies conducted or initiated by Lorus. We have initiated, are
conducting or conducted Phase I/II clinical trials of GTI-2040 in patients
with
refractory or relapsed acute myeloid leukemia (“AML”), metastatic breast cancer,
non-small cell lung cancer, solid tumors, advanced unresectable colon cancer,
hormone refractory prostate cancer, high grade myelodysplastic syndrome (“MDS”)
and acute leukemia (“AL”). Our collaboration with the NCI is active
and ongoing. In addition, the Company is pursuing a Phase II clinical trial
with
GTI-2040 and high dose Ara-C in refractory and relapsed AML and completed
a
Phase I/II study of advanced, end-stage renal cell cancer.
siRNA
As
a complement to our antisense therapy, we are exploiting RNA intereference
technology using a novel class of small interefering RNA (“siRNA”)
molecules. SiRNA has the potential to decrease the
cellular target RNA expression though a process known as RNA
interference.
GTI-2501
Our
other antisense therapy, GTI-2501, is currently in a Phase II clinical trial
for
the treatment of hormone refractory prostate cancer at the Toronto Sunnybrook
Regional Cancer Centre, following the successful conclusion of a Phase I
clinical trial in the United States.
Other
We
have also entered into a collaboration agreement in respect of our antisense
therapy, GTI-2601 and have other antisense molecules in pre-clinical
development.
Small
Molecule
We
believe we have small molecule drug screening technologies and preclinical
scientific expertise, which we are using to create a drug candidate pipeline.
Our proprietary group of novel small molecule compounds,
-
5
-
which
include lead compounds LT-253 and ML-220, have unique structures and modes
of
action, and are promising candidates for the development of novel anticancer
agents with high safety profiles. See “-- Clinical Development” and
“Business of the Company - Small Molecule Therapies”.
Immunotherapy
Lorus’
immunotherapy product candidates are Virulizin® and IL-17E. See “--
Clinical Development” and “Business of the Company - Immunotherapy” for more
details.
Virulizin®
In
2002, we initiated a phase III clinical trial of Virulizin® for patients with
locally advanced or metastatic pancreatic cancer who had not previously received
systemic chemotherapy. In July of 2005, we announced the completion
of the study and in October 2005, we announced that the results of the trial
indicated that the overall survival rate of patients who were treated with
Virulizin® plus gemcitabine (a standard chemotherapy drug) was not statistically
significant when compared to those patients in the study who were given
gemcitabine plus a placebo. Subsequent sub-group analyses support the potential
for further study in select patient populations. We are currently seeking
partners to continue the clinical development of Virulizin®.
IL-17E
We
have discovered a new lead drug candidate, IL-17E, which belongs to a larger
family of cytokines. In experiments with mice, IL-17E has demonstrated
significant antitumor activity against a variety of human tumors, including
melanoma, pancreatic, colon, lung and ovarian tumors grown in mice. We believe
that these preliminary animal results support our further investigation of
the
potential clinical applications of IL-17E.
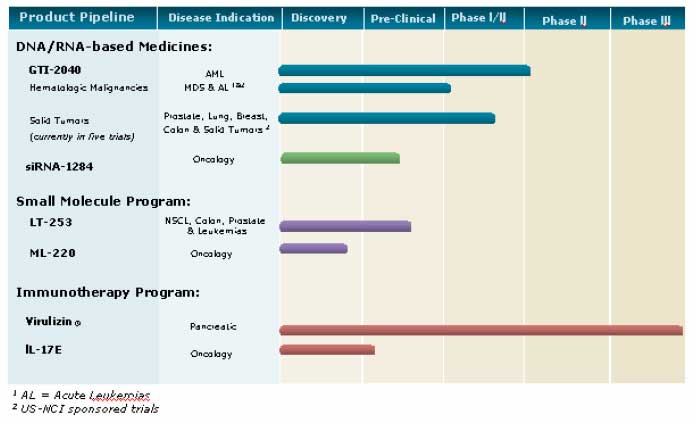
Clinical
Development
The
chart below illustrates our current view of the clinical development stage
of
each of our products. This chart reflects the current regulatory
approval process for biopharmaceuticals in Canada and the United States (with
the exception of Virulizin® for malignant melanoma which is approved for use in
the private market in Mexico). See “Regulatory Requirements” for a
description of the regulatory approval process in Canada and the United States.
These qualitative estimates of the progress of our products are intended
solely
for illustrative purposes and the information contained herein is qualified
in
its entirety by the information appearing elsewhere or incorporated by reference
in this annual information form.
-
6
-
REGULATORY
REQUIREMENTS
Overview
Regulation
by government authorities in Canada, the United States, Mexico and the European
Union is a significant factor in our current research and drug development
activities. To clinically test, manufacture and market drug products
for therapeutic use, we must satisfy the rigorous mandatory procedures and
standards established by the regulatory agencies in the countries in which
we
currently operate or intend to operate.
The
laws of most of these countries require the licensing of manufacturing
facilities, carefully controlled research and the extensive testing of
products. Biotechnology companies must establish the safety and
efficacy of their new products in clinical trials, establish cGMP and control
over marketing activities before being allowed to market their
products. The safety and efficacy of a new drug must be shown through
clinical trials of the drug carried out in accordance with the mandatory
procedures and standards established by regulatory agencies.
The
process of completing clinical trials and obtaining regulatory approval for
a
new drug takes a number of years and requires the expenditure of substantial
resources. Once a new drug or product license application is
submitted, we cannot assure you that a regulatory agency will review and
approve
the application in a timely manner. Even after initial approval has
been obtained, further studies, including post-marketing studies, may be
required to provide additional data on efficacy and safety necessary to confirm
the approved indication or to gain approval for the use of the new drug as
a
treatment for clinical indications other than those for which the new drug
was
initially tested. Also, regulatory agencies require post-marketing
surveillance programs to monitor a new drug’s side effects. Results
of post-marketing programs may limit or expand the further marketing of new
drugs. A serious safety or effectiveness problem involving an
approved new drug may result in a regulatory agency requiring withdrawal
of the
new drug from the market and possible civil action. We cannot assure
you that we will not encounter such difficulties or excessive costs in our
efforts to secure necessary approvals, which could delay or prevent us from
manufacturing or marketing our products.
-
7
-
In
addition to the regulatory product approval framework, biotechnology companies,
including Lorus, are subject to regulation under local provincial, state
and
federal law, including requirements regarding occupational safety, laboratory
practices, environmental protection and hazardous substance control, and
may be
subject to other present and future local, provincial, state, federal and
foreign regulation, including possible future regulation of the biotechnology
industry.
Canada
In
Canada, the manufacture and sale of new drugs are controlled by Health Canada
(“HC”). New drugs must pass through a number of testing stages,
including pre-clinical testing and clinical trials. Pre-clinical
testing involves testing the new drug’s chemistry, pharmacology and toxicology
in vitro and in vivo. Successful results (that is,
potentially valuable pharmacological activity combined with an acceptable
low
level of toxicity) enable the developer of the new drug to file a clinical
trial
application (“CTA”) to begin clinical trials involving humans.
To
study a drug in Canadian patients, a CTA submission must be filed with
HC. The CTA submission must contain specified information, including
the results of the pre-clinical tests completed at the time of the submission
and any available information regarding use of the drug in humans. In
addition, since the method of manufacture may affect the efficacy and safety
of
a new drug, information on manufacturing methods and standards and the stability
of the drug substance and dosage form must be presented. Production
methods and quality control procedures must be in place to ensure an acceptably
pure product, essentially free of contamination, and to ensure uniformity
with
respect to all quality aspects.
Provided
HC does not reject a CTA submission, clinical trials can
begin. Clinical trials for product candidates to treat cancer are
generally carried out in three phases. Phase I involves studies to
evaluate toxicity and ideal dose levels in humans. The new drug is
administered to human patients who have met the clinical trial entry criteria
to
determine pharmacokinetics, human tolerance and prevalence of adverse side
effects. Phases II and III involve therapeutic studies. In
Phase II, efficacy, dosage, side effects and safety are established in a
small
number of patients who have the disease or disorder that the new drug is
intended to treat. In Phase III, there are controlled clinical trials
in which the new drug is administered to a large number of patients who are
likely to receive benefit from the new drug. In Phase III, the
effectiveness of the new drug is compared to that of standard accepted methods
of treatment in order to provide sufficient data for the statistical proof
of
safety and efficacy for the new drug.
If
clinical studies establish that a new drug has value, the manufacturer submits
a
new drug submission (“NDS”) application to HC for marketing
approval. The NDS contains all information known about the new drug,
including the results of pre-clinical testing and clinical
trials. Information about a substance contained in an NDS includes
its proper name, its chemical name, and details on its method of manufacturing
and purification, and its biological, pharmacological and toxicological
properties. The NDS also provides information about the dosage form
of the new drug, including a quantitative listing of all ingredients used
in its
formulation, its method of manufacture, manufacturing facility information,
packaging and labelling, the results of stability tests, and its diagnostic
or
therapeutic claims and side effects, as well as details of the clinical trials
to support the safety and efficacy of the new drug. Furthermore, for
biological products, an on-site evaluation is required prior to the issuance
of
a notice of compliance (“NOC”). All aspects of the NDS are critically
reviewed by HC. If an NDS is found satisfactory, a NOC is issued permitting
the
new drug to be sold. In Canada an Establishment license must be
obtained prior to marketing the product.
HC
has a policy of priority evaluation of new drug submissions for all drugs
intended for serious or life-threatening diseases for which no drug product
has
received regulatory approval in Canada and for which there is reasonable
scientific evidence to indicate that the proposed new drug is safe and may
provide effective treatment.
-
8
-
The
monitoring of a new drug does not cease once it is on the market. For
example, a manufacturer of a new drug must report any new information received
concerning serious side effects, as well as the failure of the new drug to
produce desired effects. As well, if HC determines it to be in the
interest of public health, a notice of compliance for a new drug may be
suspended and the new drug may be removed from the market.
An
exception to the foregoing requirements relating to the manufacture and sale
of
a new drug is the limited authorization that may be available in respect
of the
sale of new drugs for emergency treatment. Under the special access
program, HC may authorize the sale of a quantity of a new drug for human
use to
a specific practitioner for the emergency treatment of a patient under the
practitioner’s care. Prior to authorization, the practitioner must
supply HC with information concerning the medical emergency for which the
new
drug is required, such data as is in the possession of the practitioner with
respect to the use, safety and efficacy of the new drug, the names of the
institutions at which the new drug is to be used and such other information
as
may be requested by HC. In addition, the practitioner must agree to
report to both the drug manufacturer and HC the results of the new drug’s use in
the medical emergency, including information concerning adverse reactions,
and
must account to HC for all quantities of the new drug made
available.
The
Canadian regulatory approval requirements for new drugs outlined above are
similar to those of other major pharmaceutical markets. While the
testing carried out in Canada is often acceptable for the purposes of regulatory
submissions in other countries, individual regulatory authorities may request
supplementary testing during their assessment of any submission. We cannot
assure you that the clinical testing conducted under HC authorization or
the
approval of regulatory authorities of other countries will be accepted by
regulatory authorities outside Canada or such other countries.
United
States
In
the United States, the FDA controls the manufacture and sale of new
drugs. New drugs require FDA approval of a marketing application
(e.g. an NDA or FDA application) prior to commercial
sale. To obtain marketing approval, data from adequate and
well-controlled clinical investigations, demonstrating to the FDA’s satisfaction
a new drug’s safety and effectiveness for its intended use, are
required. Such data are generated in studies conducted pursuant to an
IND submission, similar to that required for a CTA in Canada. As in
Canada, clinical studies are characterized as Phase I, Phase II and Phase
III
trials or a combination thereof. In a marketing application, the
manufacturer must also demonstrate the identity, potency, quality and purity
of
the active ingredients of the new drug involved, and the stability of those
ingredients. Further, the manufacturing facilities, equipment,
processes and quality controls for the new drug must comply with the FDA’s cGMP
regulations for drugs or biological products both in a pre-licensing inspection
before product licensing and in subsequent periodic inspections after
licensing. In the case of a biological product, an establishment
license must be obtained prior to marketing and batch releasing.
A
five-year period of market exclusivity for a drug comprising a new chemical
entity (“NCE”) is available to an applicant that succeeds in obtaining FDA
approval of a NCE, provided the active ingredient of the NCE has never before
been approved in an NDA. During this exclusivity period, the FDA may not
approve
any abbreviated application filed by another sponsor for a generic version
of
the NCE. Further, a three-year period of market exclusivity for a new use
or
indication for a previously approved drug is available to an applicant that
submits new clinical studies that are essential to support the new use or
indication. During the latter period of exclusivity, the FDA may not approve
an
abbreviated application filed by another sponsor for a generic version of
the
product for that use or indication.
The
FDA has “fast track” regulations intended to accelerate the approval process for
the development, evaluation and marketing of new drugs used to diagnose or
treat
life-threatening and severely debilitating illnesses for which no satisfactory
alternative therapies exist. “Fast track” designation affords early
interaction with the FDA in terms of protocol design and permits, although
it
does not require, the FDA to
-
9
-
issue
marketing approval after completion of Phase II clinical trials (although
the
FDA will require subsequent clinical trials or even post-approval efficacy
studies).
BUSINESS
OF THE COMPANY
Overview
Chemotherapeutic
drugs have been the predominant medical treatment option for cancer,
particularly metastatic cancer, for the past 30 years. More recently, a range
of
novel cancer drugs have been developed that are efficacious while improving
patient quality of life. Unlike chemotherapies, which are typically
based on chemical synthesis, these new drugs may be of biological origin,
based
on naturally occurring molecules, proteins or genetic material. While
chemotherapy drugs are relatively non-specific and as a result toxic to normal
cells, these biological agents specifically target individual molecules or
genes
that are involved in disease and are therefore preferentially toxic to tumor
cells. The increased specificity of these drugs may result in fewer
and milder side effects, meaning that, in theory, larger and therefore, more
effective doses can be administered. The current paradigm in cancer management
is a multi-modal approach that combines multiple treatment options tailored
to
the specific indication and individual patient. As a result, drug regimens
that
combine novel small molecule chemotherapies based on emerging understanding
of
cancer development with biological agents are of considerable
interest.
We
believe that the future of cancer treatment and management lies in drugs
that
are effective, safe and have minimal side effects leading to improved patient
quality of life. Many of the drugs currently approved for the treatment and
management of cancer are toxic resulting in severe side effects that limit
dosing and efficacy. We believe that a product development plan based
on effective and safe drugs would have broad applications in cancer treatment.
Lorus’ strategy is to continue the development of our product pipeline using
several therapeutic approaches. Each therapeutic approach is dependent on
different technologies, which we believe mitigates the development risks
associated with a single technology platform. In developing and evaluating
our
products, we evaluate the merits of each product throughout the clinical
trial
process and consider commercialization opportunities.
Antisense-DNA/RNA-based
Therapeutics
Introduction
Metabolism,
cell growth and cell division are tightly controlled by complex protein
signalling pathways in response to specific conditions, thereby maintaining
normal function. Many human diseases, including cancer, can be traced to
faulty
protein production and/or regulation. As a result, traditional therapeutics
are
designed to interact with the disease-causing proteins and modify their
function. A significant number of current anticancer drugs act by damaging
either DNA or proteins within cells (e.g., chemotherapy) or by inhibiting
the
function of proteins or small molecules (e.g. estrogen blockers, such
as Tamoxifen). Antisense therapeutics offer a novel approach to treatment
in
that they are designed to prevent the production of proteins causing
disease.
The
premise of this therapeutic approach is to target an earlier stage of the
biochemical process than is usually possible with conventional drugs. The
blueprint for protein production is encoded in the DNA of each cell. To
translate this code into protein the cell first produces mRNAs (messenger
ribonucleic acids) specific to each protein and these act as intermediaries
between the information encoded in DNA and production of the corresponding
protein. Most traditional therapies interact with the final synthesized or
processed protein. Often this interaction lacks specificity that
would allow for interaction with only the intended target, resulting in
undesired side effects. In contrast, this newer approach alters
gene-expression at the mRNA level, prior to protein synthesis, with specificity
such that expression of only the intended target is affected. We
believe that
-
10
-
drugs
based on this approach may have broad applicability, greater efficacy and
fewer
side effects than conventional drugs.
We
have developed a number of antisense drugs, of which our lead products are
GTI-2040 and GTI-2501. These products target the two components of
ribonucleotide reductase (“RNR”). RNR is a highly regulated, cell
cycle-controlled protein required for DNA synthesis and repair. RNR
is made up of two components, R1 and R2, encoded by different genes. RNR
is
essential for the formation of deoxyribonucleotides, which are the building
blocks of DNA. Since RNR activity is highly elevated in tumor cell
populations and is associated with tumor cell proliferation, we have developed
antisense molecules specific for the mRNA of the R1 (GTI-2501) or the R2
(GTI-2040) components of RNR. Furthermore, the R2 component also appears
to be a
signal molecule in cancer cells and its elevation is believed to modify a
biochemical pathway that can increase the malignant properties of tumor cells.
Consequently, reducing the expression of the RNR components in a tumor cell
with
antisense drugs is expected to have antitumor effects.
GTI-2040
Our
lead antisense therapy is GTI-2040, an antisense drug that targets the R2
component of RNR and has exhibited antitumor properties against over a dozen
different human cancers in standard mouse models, including chemotherapy
resistant tumors. We have completed a Phase I/II clinical trial of
GTI-2040 for advanced or metastatic renal cell carcinoma. We are also
conducting or have completed a multiple Phase I/II clinical trial program
in
cooperation with the NCI, for the study of GTI-2040 for the treatment of
AML,
breast cancer, lung cancer, colon cancer, prostate cancer , a series of solid
tumors and myelodysplastic syndrome and acute leukemia. We also
recently initiated Phase II clinical trial with GTI-2040 and high dose Ara-C
in
refractory and relapsed AML.
Pre-clinical
Testing
GTI-2040
has demonstrated excellent anti-tumour activity in a number of murine models
of
human cancer including xenograft tumour growth, metastasis and survival models.
The results of these studies were published in the June 1, 2003 issue of
Cancer Research. Additional studies have demonstrated combination drug
efficacy in xenograft tumour growth studies for human cancer cells, including
drug resistant tumour cell lines. More recent studies, the results of which
were
presented at the 2007 annual meeting of the AACR, focus on dose schedule
optimization for GTI-2040 in combination with docetaxel. These studies
demonstrate that the timing of these two drugs can be optimized: observations
that have implications for the ongoing NCI sponsored clinical trials. These
studies continued in 2007. Lorus has also published results from studies
aimed
at development of an assay for R2 determination from clinical samples
(Journal of Clinical Laboratory Analysis, 2005). Formal pre-clinical
development of GTI-2040, including manufacturing and toxicology studies,
was
initiated in mid-1998. Pre-clinical studies, including GLP toxicology
studies in standard animal models, have demonstrated that GTI-2040 is well
tolerated at concentrations that exceed commensurate therapeutic doses in
humans.
Clinical
Development
Lorus
Sponsored Trials
Acute
Myeloid Leukemia:
In
August 2007, we announced an expansion of GTI-2040 development program in
AML
indication with initiation of a more advanced Phase II clinical trial with
GTI-2040 and high dose Ara-C in refractory (HiDAC) and relapsed AML. This
Phase
II study includes both an efficacy study and a novel additional study to
measure
intracellular target activities and pharmacological synergies between the
two
agents. In the first stage of the 60 patient trial, the pharmacologic and
target
related activity of GTI-2040 and HiDAC will be
-
11
-
evaluated
in two groups, to determine the contribution of each agent alone and in
combination. The second stage of the trial will provide efficacy evaluation
in a
larger patient population. Lorus expects the clinical trial to be completed
by
the end of 2008. The decision to advance clinical development of GTI-2040
is
based on the encouraging results from our recently completed proof of concept
NCI-sponsored study of GTI-2040 in combination with HiDAC in patients with
refractory and relapsed AML.
Advanced
Renal Cell Cancer:
In
April 2005, we announced completion of a Phase I/II clinical trial of GTI-2040
in combination with capecitabine, in patients with advanced, end-stage renal
cell cancer in the United States. This trial was a single-arm pilot study
examining the safety and efficacy of GTI-2040 used in combination with the
anticancer agent capecitabine. The majority of patients had failed two or
more
prior therapies before entering the study, exhibited extensive metastases,
and
were representative of a population with very poor prognostic outcome in
renal
cell cancer. All 33 patients entering this study had advanced disease
with multiple metastatic sites, with or without prior removal of the primary
kidney tumor. However, more than half (52%) of the patients on the recommended
dose exhibited disease stabilization or better, including one confirmed partial
response. GTI-2040 was well tolerated when combined with a cytotoxic agent
with
expected adverse events. The results of this study were accepted for publication
in the journal Cancer Chemotherapy and Pharmacology in 2007 (June 2007
e-pub date). Lorus is actively searching for partnerships
to assist with the further development of GTI-2040 for the treatment of renal
cell cancer.
NCI
Sponsored Trials
Current
clinical development for GTI-2040 is in conjunction with the US NCI, which
pays
for the cost of all clinical trials. See “-- Agreements -
Collaboration Agreements - National Cancer Institute”. To date we
have announced and/or initiated seven clinical trials with the NCI for GTI-2040
in patients with AML, metastic breast cancer, non-small all lung cancer,
solid
tumors, unresectable colon cancer, hormone refractory prostate cancer, and
MDS
and acute leukemia. These indications were selected based on the most
promising results from our preclinical studies. Upon receipt of the clinical
data from the ongoing NCI clinical trials, Lorus will
analyze and make decisions regarding the strategic direction of our antisense
portfolio. Lorus continues to search for partnerships for the future
development of GTI-2040.
In
September 2005, Lorus announced a steering committee assessment of progress
in
the ongoing U.S. NCI-sponsored clinical studies of GTI-2040. The committee
concluded that all six studies continue to progress without unacceptable
toxicity. Studies reviewed in this process included GTI-2040 in combination
with
chemotherapies in non-small cell lung cancer (NSCLC), hormone refractory
prostate cancer (HRPC), breast cancer, acute myeloid leukemia (AML), colorectal
cancer and a variety of solid tumors. Combination chemotherapies under study
include docetaxel, capecitabine, oxaliplatin, cytarabine, and
gemcitabine.
Acute
Myeloid Leukemia:
In
July 2003, we announced the FDA’s approval of the NCI-sponsored IND application
for a clinical trial of GTI-2040 in combination with cytarabine, in patients
with refractory or relapsed AML. Cytarabine is the current established drug
for
treating AML patients. The study is part of a Phase II clinical
program to be conducted under the sponsorship of the Cancer Treatment Evaluation
Program of the NCI pursuant to a clinical trial agreement between Lorus and
the
NCI.
In
December 2005, we announced interim data from the NCI-sponsored trial of
GTI-2040 in acute myeloid leukemia. The data presented showed
complete responses in 44 per cent of patients 60 years of age or
younger. Patients in this trial had either failed to respond to prior
therapy or had rapidly relapsed and as such had a low expectation of response
to
subsequent treatment (10-20%). Complete responses in the clinical
trial directly correlated with down regulation of R2, the intracellular target
of GTI-2040, demonstrating drug specificity and providing strong evidence
for an
antisense mechanism of action. Toxicities for the
-
12
-
combination
were comparable to those expected for cytarabine alone and were non
dose-limiting. Updated results were presented at the 2006 annual
meeting of ASCO and support the continued dose escalation study in younger
cohorts of patients to establish a recommended phase II dose. The AML study
group developed a novel method for analysis of GTI-2040 in biological samples
(2006 issue of Pharmaceutical Research). Furthermore this group has
reported the results of metabolic and pharmacokinetic analyses at the annual
meeting of the American Association of Pharmaceutical Scientists and the
2006
meeting of the International Society of Xenobiotics. Results have also been
published in volume 8, issue 4 of the American Association of Pharmaceutical
Scientists Journal. These studies demonstrate the uptake and accumulation
of GTI-2040 in target tissues, important observations in support of an antisense
mechanism of action for this drug candidate.
In
August 2007, we announced the completion of this study. This clinical trial
demonstrated safety and appropriate dosing of the combination regimen and
showed
promising clinical responses in patients under 60 years of
age. Moreover, the clinical responses correlated with downregulation
of R2, the cellular target of GTI-2040, and were further supported by
demonstration of intracellular GTI-2040 in circulating and bone marrow leukemic
cells. Complete results from the clinical trial are expected to be presented
by
the investigators in a scientific publication.
Metastatic
Breast Cancer:
In
August 2003, we announced that the FDA had approved the NCI’s IND to begin a
Phase I/II clinical trial to investigate GTI-2040 as a treatment for metastatic
breast cancer in combination with capecitabine (Xeloda, manufactured by Roche
Laboratories Inc.). In support of continued studies aimed at demonstrating
R2
target down-regulation in patient samples this group, in collaboration with
Lorus, published preliminary results of RT-PCR studies in the May issue of
Oncology Reports. The results demonstrate that the assay
developed by Lorus can feasibly assess R2 level is blood and tumour tissues
from
patients before and after treatment. This study is
ongoing.
Non-Small
Cell Lung Cancer:
In
September 2003, we received approval from Health Canada for initiation of
a
clinical trial of GTI-2040 in combination with docetaxel for the treatment
of
advanced non-small cell lung cancer (“NSCLC”), as part of a Phase I/II clinical
program of GTI-2040 in collaboration with the NCI. Interim results from this
study were announced in May 2005. Our interim results showed that the toxicity
profile was determined to be acceptable for the specific combination therapy
and
the observed level of disease stabilizations was encouraging given the advanced
stage of the disease in this subset of patients. The study group published
a
paper in the December issue of the Journal of Chromatography,
outlining the development of a method for determination of GTI-2040
in
human plasma samples. This highly sensitive method will be used for
pharmacokinetic studies in patient samples from the trial. This study
is ongoing.
Solid
Tumors:
In
February 2004, we announced the initiation of a Phase I/II clinical trial
examining the use of GTI-2040 in combination with gemcitabine in patients
with
solid tumors. In June 2005, results from the trial were
published. The trial was intended to identify the recommended dose of
GTI-2040 and its toxicity profile. At the recommended dose GTI-2040
demonstrated a manageable toxicity profile and was generally well tolerated
when
given as a single agent. This study is ongoing.
Unresectable
Colon Cancer:
In
May 2004, we announced the initiation of a Phase I/II clinical trial examining
GTI-2040 in combination with oxaliplatin and capecitabine in the treatment
of
advanced unresectable colon cancer. This study is part of a clinical
trials program sponsored by the NCI. This study is
ongoing.
-
13
-
Hormone
Refractory Prostate Cancer:
In
November 2004, we announced the initiation of a Phase I/II clinical trial
examining GTI-2040 in combination with docetaxel and prednisone in hormone
refractory prostate cancer. In November 2005, we announced interim
data from this trial. The data showed that along with an acceptable
tolerability profile, nine of 22 PSA evaluable patients demonstrated a PSA
response (reductions of greater than 50%). PSA is overproduced in
prostate cancer cells and is commonly used to assess disease progression
and
response. This data was also presented at the 2006 annual meeting of
ASCO.
High
Grade Myelodysplastic Syndrome and acute leukemia:
Lorus
announced in June 2006 a plan for a new clinical investigation of GTI-2040
as a
single-agent in patients with high grade myelodysplastic syndrome and acute
leukemia. This trial was initiated in mid 2007. This clinical study is designed
to evaluate the safety and activity of GTI-2040 as a single agent for acute
leukemia and MDS using a novel treatment schedule. The effect on leukemic
blasts
and blood count recovery will be assessed as part of a detailed investigation
of
the pharmacodynamic and pharmacokinetic effects, dose-response
relationships and tolerability of GTI-2040 during multiple courses of
treatment.
Orphan
Drug Status
On
March 12, 2003, the FDA awarded Orphan Drug Status to GTI-2040 for the treatment
of renal cell carcinoma.In May 2005, Lorus received Orphan Drug designation
from
the FDA for GTI-2040 in the treatment of AML.
siRNA
In
2003, Lorus began development of an anticancer therapeutic based on
siRNA-mediated inhibition of R2 expression. Early screening experiments have
identified lead compounds and preliminary in vitro and in vivo
characterization of these compounds has yielded promising
results. The
results of these studies were published in the April 2007 issue of
Anti-Cancer Drugs and were presented at the 2007 annual meeting of the
AACR. siRNA-1284, the lead compound identified from the screening study,
specifically targets R2 expression. In in vitro studies,
down-regulation of R2 expression by siRNA-1284 results in
decreased tumor cell growth (proliferation) with a concomitant
block in cell cycle progression. Furthermore, siRNA-1284 demonstrates anti-tumor
activity against human kidney, skin and colon cancer in mouse experimental
models of tumor growth. We feel that the results of these studies warrant
further development of siRNA-1284 as well as expansion of siRNA research
to
other cancer targets.
GTI-2501
Our
other antisense therapy currently in clinical development is GTI-2501. GTI-2501
targets the R1 subunit of RNR and has been shown to have antitumor activity
and
a good safety profile in pre-clinical testing. A Phase I trial also
demonstrated the safety of GTI-2501.
Pre-clinical
Testing
GTI-2501
has demonstrated antitumor activity in a number of standard mouse models
of
cancer progression including xenografts tumour growth, metastasis and survival
models. GTI-2501 was effective against a broad range of cancers including
human
breast, kidney and prostate cancers (Results published in the February 2006
Issue of the International Journal of Oncology). In
addition, pre-clinical studies have demonstrated that GTI-2501 is well tolerated
in standard animal models at concentrations that exceed commensurate therapeutic
doses in humans.
-
14
-
Clinical
Development Program
GLP-toxicology
studies for GTI-2501 were completed in November 2000 and approval of an IND
was
received from the FDA in February 2001. A Phase I dose-escalating study at
the
University of Chicago Medical Center was designed to establish the recommended
clinical Phase II dose as well as look at the safety profile of
GTI-2501. A total of 34 patients with solid tumors or lymphoma were
enrolled and have been evaluated following clinical completion. The study
demonstrated a reasonable safety profile for GTI-2501 up to the predicted
therapeutically relevant dose. In December 2003, we announced that a Phase
I/II
clinical trial for the treatment of hormone refractory prostate cancer (HRPC)
had been initiated at the Toronto Sunnybrook Regional Cancer Centre, in which
GTI-2501 is administered in combination with docetaxel. The combination of
GTI-2501 and docetaxel in this clinical trial is being investigated in patients
with asymptomatic or symptomatic HRPC where disease progression is
uncontrolled. This represents the first clinical trial of GTI-2501 in
Canada following the successful conclusion of the Phase I clinical trial
in 2004
in the United States. We announced expansion of this ongoing HRPC
trial to two additional sites in Canada in July 2004. The results of the
dose
escalation portion of the trial were presented at the 2006 annual meeteing
of
ASCO. This portion of the trial involved 13 patients in three dose cohorts.
The
results demonstrated that GTI-2501 given in combination with docetaxel was
safe
at the highest dose of GTI-2501 planned. These results warranted initiation
of
the Phase II portion of the trial which is currently ongoing.
GTI-2601
GTI-2601
is an antisense compound that targets thioredoxin, a gene whose increased
expression has been implicated in cancer progression and poor prognosis.
GTI-2601 is an effective anti-cancer agent in pre-clinical studies in animal
models of human colon cancer. The results of these studies were published
in the
February 2006 issue of Anti-cancer Drugs.
On
April 5, 2005 we announced that we had signed a collaboration agreement with
one
of Japan’s leading pharmaceutical companies, Sumitomo Pharmaceuticals Co. Ltd.
(“Sumitomo”) and Koken Co. Ltd (“Koken”) with respect to GTI-2601, our lead
antisense compound targeting thioredoxin, a gene that is over-expressed in
many
tumor tissues and has been correlated with poor prognosis and chemotherapy
resistance. Sumitomo and Koken have developed an advanced delivery
system based on collagen complexed with macromolecules. The
collaboration agreement provides that Sumitomo and Koken will further develop
their delivery technology to complex with GTI-2601, so that increased efficacy
is provided with decreased doses of the antisense drug. This
agreement provides that Lorus, Sumitomo and Koken will jointly own the compounds
that result from this collaboration (Lorus will share the results of the
collaboration with Sumitomo and Koken, 1:1).
Other
Antisense Targets
Lorus’
antisense
technology platform
extends further to other anti-cancer drug targets including thioredoxin,
thioredoxin reductase, neuropilin/VEGF165R and insulin-like
growth factor II (IGF-II). All targets have been implicated in cancer as
a
growth stimulator, a growth factor, an inhibitor of apoptosis and/or an
angiogenic factor. These projects are in the research phase of development
which
includes screening, lead candidate identification and efficacy
studies.
Small
Molecule Therapies
Introduction
Most
anticancer chemotherapeutic treatments are DNA damaging, cytotoxic agents,
designed to act on rapidly dividing cells. Treatment with these drugs
typically includes unpleasant or even serious side effects due to
-
15
-
the
inability of these drugs to differentiate between normal and cancer cells
and/or
due to a lack of high specificity for the targeted protein. In
addition, these drugs often lead to the development of tumor-acquired drug
resistance. As a result of these limitations, a need exists for more effective
anticancer drugs. One approach is to develop small molecules with a
greater specificity as anticancer drugs. Chemical compounds weighing
less than 1000 daltons (a unit of molecular weight) are designated as small
or
low molecular weight molecules. These molecules can be designed to target
specific proteins or receptors that are known to be involved with
disease.
LT-253
In
August 2005 Lorus announced the selection of two leading small molecule
compounds from a series of novel small molecules discovered by Lorus scientists
that exhibit potent anticancer activity in in vitro screens. The
results of characterization studies of these compounds were presented at
the
2006 annual meeting of the AACR and early formulation studies were published
in
the September 2006 issue of Cancer Chemotherapy and Pharmacology .Our
studies identify the main mechanism of action of these compounds, which involves
the induction of the tumor suppressor Krüppel-like factor 4. The down regulation
of Krüppel-like factor 4 is believed to be critical in the development and
progression of certain types of cancer and presents the possibility of
exploiting a novel anticancer mechanism of action. From these two
compounds, LT-253 was selected as the lead compound for development as a
drug
candidate for the treatment of colon carcinoma and non-small cell lung cancer.
This decision was based on its potent in vitro anti-proliferative
activity, its efficacy in in vivo xenograft models of human colon and
lung cancer, and on its safety profile. Manufacturing of a GMP product,
formulation development as well as formal toxicology studies in different
animal
species with the aim of filing an IND application for the initiation of a
Phase
I clinical trial are in progress.
Other
Small Molecule Targets
Lorus
is also pursuing other candidates at earlier stages of development. These
include:
|
|
•
|
LT-253
second generation derivatives for oral
administration:
|
Further
structural modifications of LT-253 produced derivatives optimized for oral
absorption. Animal efficacy studies are in progress.
|
|
•
|
ML-220
platform
|
Lorus
is developing novel derivatives that target cancer relevant genes, which
are
critical in a major signaling pathway involved in tumorigenesis and represent
important new cancer targets. Lead optimization of ML-220 yielded several
novel
derivatives that showed potent target inhibitory activity in vitro and
in cancer cells, and growth inhibitory activity against prostate and renal
carcinoma cell lines.
Immunotherapy
Introduction
Immunotherapy
is a form of treatment that stimulates the body’s immune system to fight
diseases including cancer. Immunotherapy may help the immune system
to fight cancer by improving recognition of differences between healthy cells
and cancer cells. Alternatively it may stimulate the production of specific
cancer fighting cells.
-
16
-
Virulizin®
Virulizin®,
Lorus’ immunotherapeutic drug, has been shown in pre-clinical studies to be an
effective immunotherapy that stimulates monocytes and macrophages to infiltrate
tumor tissue and attack tumor cells. The ability to stimulate NK cells and
macrophages results in Virulizin® anti-tumour efficacy demonstrated in a number
of animal models of human tumour growth. Monocytes and macrophages
are types of white blood cells that are key players in the immune response
to
foreign pathogens and tumor cells. When macrophages and monocytes are
activated, they produce proteins called cytokines that have the ability to
kill
tumor cells directly. Our studies indicate that Virulizin® stimulates
the release of tumor necrosis factor (TNF-alpha), one type of cytokine, in
immune cells to induce apoptosis (programmed cell death) of tumor cells.
In
addition, Virulizin® has been shown to increase the expression of IL-12 in
macrophages. The resulting increased levels of IL12 in mouse serum
lead to NK cell activation. Since 2003 the results of these studies have
been
published in five peer-reviewed scientific journals and presented at a number
of
international conferences.
Our
studies indicate that Virulizin® produces fewer negative side effects than
commonly used chemotherapy agents likely because the drug works by stimulating
the immune system to attack the cancer, rather than directly killing cancerous
cells.
Clinical
Development Program
In
2002 Lorus initiated a Phase III double-blind, multicenter, randomized study
in
patients with locally advanced or metastatic pancreatic cancer who had not
previously received systemic chemotherapy. This clinical trial was conducted
at
over 100 sites in North America and Europe with enrolment of 436 patients
with
advanced pancreatic cancer. Patients enrolled in the study were
randomly selected to receive treatment with either: (i) Virulizin® plus
gemcitabine or (ii) placebo plus gemcitabine. Optional second line therapy
for
those patients who failed to respond or became resistant to gemcitabine included
Virulizin® or placebo, alone or in combination with 5-fluorouracil
(“5-FU”). All study subjects were monitored throughout the remainder
of their lifespan. The end points of the study were survival and
clinical benefits. In July 2005 Lorus announced completion of “last patient
visit” for the phase III trial. Lorus announced the results of the
phase III trial in October 2005 and those results are discussed in detail
below.
Clinical
Trial Results
In
October 2005, we released the results of the Phase III clinical trial evaluating
Virulizin® for the treatment of pancreatic cancer. The primary end
points of the study were not met. For the efficacy evaluable
population, the study showed that the addition of Virulizin® to gemcitabine
resulted in a median overall survival of 6.8 months and a one-year survival
rate
of 27.2%, compared to 6.0 months and 16.8% for placebo plus
gemcitabine. In the intent to treat population the median overall
survivals were 6.3 months for Virulizin plus gemcitabine (one year survival
rate
of 25.9%) compared to 6.0 months for placebo plus gemcitabine (one year survival
rate of 17.6%). While comparison of the median overall survival times
did not reach statistical significance, exploratory analysis did show promising
trends in specific patient populations. The results of the exploratory sub-group
analyses were presented at the 2006 annual meeting of the American Society
of
Clinical Oncology (“ASCO”). From these analyses the following sub-groups were
identified as having demonstrated benefit that approaches statistical
significance: patients with low ECOG scores (better overall performance),
patients with metastatic disease and patients that continued Virulizin® therapy
during second line therapy. In addition, those patients that continued
Virulizin® during salvage therapy demonstrated a survival benefit that was
statistically significant.
Lorus
is currently seeking partners to continue the clinical development of Virulizin®
in these patient specific populations.
-
17
-
Orphan
Drug
Lorus
received Orphan Drug designation from the United States Food and Drug
Administration (“FDA”) in February 2001 for Virulizin® in the treatment of
pancreatic cancer. Orphan drug status is awarded to drugs used in the
treatment of a disease that afflicts less than 200,000 patients annually
in the
United States to encourage research and testing. This status means
that the FDA will help to facilitate the drug’s development process by providing
financial incentives and granting seven years of market exclusivity in the
United States (independent of patent protection) upon approval of the drug
in
the United States. In June 2005, Lorus announced that Virulizin® was granted
Orphan Drug status in the European Union for pancreatic cancer.
IL-17E
Lorus
has recently discovered a new lead drug candidate, IL-17E, which belongs
to a
larger family of cytokines. The results of these studies were presented at
the
2006 annual meeting of the American Association for Cancer Research (“AACR”).
IL-17E has demonstrated significant antitumor activity against a variety
of
human tumors, including melanoma, pancreatic, colon, lung and ovarian tumors
grown in mice. In addition, combinations of IL-17E with chemotherapeutic
agents
showed enhanced anti-tumor efficacy against human colon, lung, melanoma and
ovarian tumor models in mice. The anti-tumor activity was dose-dependent
and was
observed using three different routes of administration. Studies on the
mechanism of action showed that treatment with IL-17E resulted in increased
serum levels of IL-5 and increased percentages of eosinophils in peripheral
blood. Spleen cells isolated from IL-17E-treated mice showed increases in
eosinophils and B-cells, as well as an increase in the percentage of activated
B
cells. Furthermore, treatment with IL-17E resulted in phosphorylation of
kinases
and activation of transcription factors involved in immune stimulation. Taken
together, the data support further investigation of the potential clinical
application of IL-17E, placing IL-17E in a growing class of anticancer
immunotherapeutic drugs.
Other
Technologies
We
are currently assessing several new technologies for their potential as new
drug
candidates. They include technologies in areas of tumor suppressor
gene therapy and other small molecule technology platform that we believe
to
have the potential to work through a unique mechanism of action to decrease
the
expression of cancer relevant genes.
Gene
Therapy
Researchers
at Lorus have developed a gene therapy product using the R1 gene of
ribonucleotide reductase (which has been shown to act as a tumour suppressor
gene) encoded in a modified adenoviral vector (rAd5-R1) for the potential
treatment of patients with colon cancer. This project is in the pre-clinical
phase of development and has resulted in publication of an article in the
October 2003 issue of Clinical Cancer Research.
Agreements
Manufacturing
Agreements
Bio
Vectra dcl
In
July 2004, we entered into negotiations with Diagnostics Chemicals Limited
(doing business as BioVectra dcl) in Prince Edward Island for the commercial
manufacture of Virulizin®, for which a contract was
-
18
-
executed
in October 2004. BioVectra has a cGMP facility capable of large-scale commercial
production. In June 2005 Lorus announced that BioVectra had successfully
produced Virulizin® in both optimized clinical and commercial batch
scales. The contract remains in force, although Bio Vectra is not
currently performing any manufacturing of Virulizin®.
Licence
Agreements
Ion
Pharmaceuticals and Cyclacel
In
December 1997, Lorus, through NuChem, acquired certain patent rights and
a
sublicense from Ion to develop and commercialize the anticancer applications
of
CLT and new chemical entities related to CLT (the “NuChem
Analogs”). To July 2006, NuChem had made cash payments totalling US
$500,000 to Ion. The balance is payable upon the achievement of
certain milestones based on the commencement and completion of clinical trials
related to the NuChem Analogs.
The
NuChem Analog patents are ancillary to the Company’s primary development
activities and do not relate to the Company’s core research and development
focus, namely GTI-2040, nor did they relate specifically to the development
of
the Virulizin product. In addition to the amounts previously paid in
cash or shares, the Company is required to make future cash payments based
on
achieving certain future milestones on the first of any Sublicense Product
or
Lead Compound (as defined in the agreements), including: US$250 thousand
on
completion of a Phase I trial, US$500 thousand on completion of a Phase II
trial, US$750 thousand upon completion of the first Phase III trial and US$1.5
million on marketing approval for the production the United States, Canada,
England or France. The company does not currently expect to achieve
any of the above milestones in fiscal years ended May 31, 2007 or 2008 and
cannot reasonably predict when such milestones will be achieved, if at
all.
All
research and development activities to be undertaken by NuChem are to be
funded
by us through subscriptions for non-participating preference shares of
NuChem. As at May 31, 2007, we had provided a total of $5,749,000 of
funding to NuChem.
In
September 2003, Lorus, NuChem and Cyclacel Limited signed an exclusive worldwide
license agreement for the development and commercialization of the NuChem
Analogs. Under the terms of the agreement, Lorus received upfront
fees of US $400,000 and will receive milestone payments which, assuming all
milestones are achieved, will total approximately US $11.6 million for our
pre-clinical compound NC 381, and similar milestone payments for each of
any
other compounds developed from the compound library. In addition to
these payments, we will receive royalties based on product
sales. Cyclacel is responsible for all future drug development
costs.
In
reference to the Cyclacel agreement, the Company is entitled to receive certain
future milestone payments based on the commencement of future trials in relation
to those products developed by Cyclacel under the agreement including for
the
first product/follow-on products, as defined in the agreement and in certain
cases, back-up product as defined in the agreement: $US600,000 upon
commencement of a Phase II trial, US$3,000,000 on commencement of a Phase
III
trial, and between US$1,750,000 and $4,000,000 upon receipt of
marketing approval in each of various geographic areas. Thereafter
the company is entitled to a royalty of between 2.0% and 4.0% depending upon
the
level of sales. The agreement also contains certain milestone and
royalty obligations based on whether Cyclacel chooses to sublicense any of
the
products covered by that agreement. The company does not
currently expect Cyclacel to achieve any of the above milestones in fiscal
years
ended May 31, 2007 or 2008 and cannot reasonably predict when such milestones
will be achieved, if at all.
-
19
-
University
of Manitoba
The
University of Manitoba (the “University”), Dr. Jim Wright, Dr. Aiping Young and
Cancer Care entered into an exclusive license agreement (the “License
Agreement”) with GeneSense dated June 20, 1997 pursuant to which GeneSense was
granted an exclusive worldwide license to certain patent rights with the
right
to sub-license. In consideration for the exclusive license to
GeneSense of the patent rights, the University and Cancer Care are entitled
to
an aggregate of 1.67% of the net sales received by GeneSense from the sale
of
products or processes derived from the patent rights and 1.67% of all monies
received by GeneSense from sub-licenses of the patent rights. GeneSense is
solely responsible for the preparation, filing, prosecution and maintenance
of
all patent applications and patents included in the patent rights and all
related expenses. Pursuant to the terms of the License Agreement, any
and all improvements to any of the patent rights derived in whole or in part
by
GeneSense after the date of the License Agreement are not included within
the
scope of the License Agreement and do not trigger any payment of
royalties.
The
University of Manitoba agreement relates specifically to antisense patents
in
existence or pending at the time of the agreement, subsequent patent amendments
or advancements to these patents remain as the property of Lorus, without
license rights accruing back to the University of Manitoba. The
Company is currently pursing its antisense development program, primarily
as a
function of advancements and amendments to the original patents. The company
has
not yet earned any revenue from the products covered under the agreement
and
therefore has not paid any royalties under this agreement and cannot reasonably
predict the timing and amount of any future payment. The company does
not expect to make any royalty payments under this agreement in fiscal years
ended May 31, 2007 or 2008, and cannot reasonably predict when such royalties
will become payable, if at all.
Collaboration
Agreements
National
Cancer Institute
In
February 2003, Lorus and the United States National Cancer Institute approved
clinical protocols to conduct a series of clinical trials in a Phase II program
to investigate the safety and efficacy of our lead antisense drug, GTI-2040
in
breast cancer, colon cancer, non-small cell lung cancer, acute myeloid leukemia,
prostate cancer, and in a range of solid tumours. Lorus and the NCI signed
a
formal clinical trial agreement (expiring in October 2007) in which the NCI
financially sponsors the GTI-2040 clinical trials, while Lorus provides the
clinical trial drug. All six trials were in progress as of May 31,
2006. In July 2006, we announced a seventh trial to be conducted with
the NCI for GTI-2040 for the treatment of MDS and AML.
NCI
carries out clinical trials on behalf of the Company at its own
cost. The rights to publish data remains with the NCI sponsored
investigator generating the information. The commercial results of
the studies, including commercialization of any products remain with Lorus
with
no financial, license, or intellectual property rights accruing to the
Investigator or NCI for their participation.
All
projects underway are and at various stages of completion. NCI has no
rights to exploit the research results, except through the right of
investigators to publish data accumulated by it during the testing, nor does
it
have any obligation to pay or receive royalties under the
agreement. Any royalty rights on products derived from the work
performed by NCI will need to be negotiated by Lorus under a marketing agreement
with third parties (if not carried out by Lorus). It is not
possible to reasonably estimate the amount and timing of any royalty receipts,
if any.
In
regards to future payment obligations, Lorus’ obligations under this agreement
are limited to the supply of drugs, the cost for which has been
incurred. The company does not currently expect any significant costs
associated with the supply of the drug in the future, depending on the outcome
of the projects.
-
20
-
Sumitomo
and Koken
In
April 2005, we signed a collaboration agreement with Sumitomo and Koken with
respect to GTI-2601, our antisense compound targeting
thioredoxin. Sumitomo and Koken have developed an advanced delivery
system based on collagen complexed with macromolecules. The
collaboration agreement provides that Sumitomo and Koken will further develop
their delivery technology to complex with GTI-2601, so that increased efficacy
is provided with decreased doses of the antisense drug. This
agreement provides that Lorus, Sumitomo and Koken will jointly own the compounds
that result from this collaboration (Lorus: Sumitomo and Koken,
1:1).
The
company does not have any significant payment obligations for this
project. Both Lorus and Sumitomo and Koken are responsible for their
own costs during the feasibility study phase. To date the
project has not produced significant results and therefore the Company cannot
predict any royalty revenue, if any.
Other
From
time to time, we enter into other research and technology agreements with
third
parties under which research is conducted and monies expended. These
agreements outline the responsibilities of each participant and the appropriate
arrangements in the event the research produces a product
candidate.
We
also have licensing agreements to use proprietary technology of third parties
in
relation to our research and development. If this research ultimately
results in a commercialized product, we have agreed to pay certain royalties
and
licensing fees.
Business
Strategy
By
developing cancer therapeutics using different mechanisms of action that
may be
efficacious against a wide variety of cancers, we seek to maximize our
opportunity to address multiple cancer therapeutic markets. In our
efforts to obtain the greatest return on our investment in each drug candidate,
we separately evaluate the merits of each candidate throughout the clinical
trial process and consider commercialization opportunities when
appropriate. In the next fiscal year, we intend to pursue
partnerships and further development of our lead technologies.
Our
objective is to maximize the therapeutic value and potential commercial success
of GTI-2040 and the small molecule platform while at the same time pursuing
partnership opportunities for development of our immunotherapy products and
others. In the near term, we intend to pursue research and early
clinical development with our own funds with respect to GTI-2040 and the
small
molecule platform. In our efforts to obtain the greatest return on our
investment in each drug candidate, we separately evaluate the merits of each
candidate throughout the clinical trial process and will consider
commercialization opportunities when appropriate.
Financial
Strategy
To
meet future financing requirements, we intend to finance our operations through
some or all of the following methods: public or private equity or debt
financings, capital leases, and collaborative and licensing
agreements. We intend to pursue financing opportunities as they
arise.
Secured
Convertible Debentures
On
October 6, 2004, the Company entered into a Subscription Agreement (the
“Agreement”) with The Erin Mills Investment Corporation (“TEMIC”) to issue an
aggregate of $15 million of secured convertible
-
21
-
debentures
(the “Debentures”) issuable in three tranches of $5 million each, in each of,
October 2004, January 2005 and April 2005. The Debentures are secured
by a first charge over all of the assets of the Company. All
Debentures issued under the Agreement are due on October 6, 2009 and are
subject
to interest payable monthly at a rate of prime plus 1% until such time as
the
Company’s share price reaches $1.75 for 60 consecutive trading days, at which
time interest will no longer be charged. Interest is payable in
common shares of Lorus until Lorus’ shares trade at a price of $1.00 or more
after which interest will be payable in cash or common shares at the option
of
the debenture holder. Common shares issued in payment of interest are issued
at
a price equal to the weighted average trading price of such shares for the
ten
trading days immediately preceding their issue in respect of each interest
payment. The $15.0 million principal amount of Debentures is convertible
at the
holder’s option at any time into common shares of the Company with a conversion
price per share of $1.00. With the issuance of each $5.0 million
debenture, the Company issued to the debt holder 1,000,000 warrants with
a term
of five years to purchase common shares of the Company at a price per share
equal to $1.00.
As
a condition to agreeing to the Arrangement (as discussed below), the holder
of
Lorus’ $15.0 million secured convertible debenture required the repurchase by
Lorus of its outstanding three million common share purchase warrants at
a
purchase price of $252,000.
Share
Issuances
On
July 13, 2006 the company entered into an agreement with High Tech Beteiligungen
GmbH & Co. KG (“High Tech”) to issue 28.8 million common shares at $0.36 per
share for gross proceeds of $10.4 million. The subscription price represented
a
premium of 7.5% over the closing price of the common shares on the Toronto
Stock
Exchange on July 13, 2007. The closing of the transaction is subject to certain
conditions, including the approval of the Toronto Stock Exchange and the
American Stock Exchange and the filing and clearance of a prospectus in Ontario
qualifying the issuance of the common shares. The transaction closed
on August 31, 2006. In connection with the transaction, High Tech received
demand registration rights that will enable High Tech to request the
registration or qualification of the common shares for resale in the United
States and Canada, subject to certain restrictions. These demand registration
rights expire on June 30, 2012. In addition, High Tech received the right
to
nominate one nominee to the board of directors of Lorus or, if it does not
have
a nominee, it will have the right to appoint an observer to the board. Upon
completion of the transaction, High Tech held approximately 14% of the issued
and outstanding common shares of Lorus Therapeutics Inc.
On
July 24, 2007 Lorus entered into an agreement with Technifund Inc. to issue
on a
private placement basis, 5 million common shares at $0.36 per share for gross
proceeds of $1.8 million. The transaction closed on September 1,
2006.
Plan
of Arrangement and Corporate Reorganization
On
July 10, 2007, Old Lorus and the Company completed a plan of arrangement
and
corporate reorganization with, among others, 6707157 Canada Inc. (“Investor’)
and Pinnacle International Lands, Inc. (the “Arrangement”). As part
of the Arrangement, all of the assets and liabilities of Old Lorus (including
all of the shares of its subsidiaries held by it), with the exception of
certain
future tax assets were transferred, directly or indirectly, from Old Lorus
to
the Company. Securityholders in Old Lorus exchanged their securities
in Old Lorus for equivalent securities in New Lorus (the "Exchange") and
the
board of directors and management of Old Lorus continued as the board of
directors and management of New Lorus. New Lorus obtained
substitutional listings of its common shares on both the Toronto Stock Exchange
and the American Stock Exchange.
-
22
-
As
part of the Arrangement, the Company changed its name to Lorus Therapeutics
Inc.
and continues as a biopharmaceutical company, specializing in the research
and
development of pharmaceutical products and technologies for the management
of
cancer as a continuation of the business of Old Lorus.
In
connection with the Arrangement and after the Exchange, the share capital
of Old
Lorus was reorganized into voting common shares and non-voting common shares
and
Investor acquired from New Lorus and Selling Shareholders (as defined below)
approximately 41% of the voting common shares and all of the non-voting common
shares of Old Lorus for a cash consideration of approximately $8.5 million
on
closing of the transaction less an escrowed amount of $600,000, subject to
certain post-closing adjustments and before transaction costs. The
remaining 59% of the voting common shares of Old Lorus were distributed to
the
shareholders of New Lorus who were not residents of the United States on
a
pro-rata basis. Shareholders of New Lorus who were residents of the
United States received a nominal cash payment in lieu of their pro-rata share
of
voting common shares of Old Lorus. After completion of the
Arrangement, New Lorus is not related to the former Lorus Therapeutics Inc.,
which was subsequently renamed 4325231 Canada Inc.
As
a condition of the Arrangement, High Tech Beteilingungen GmbH & Co. KG and
certain other shareholders of Old Lorus (the “Selling Shareholders”) agreed to
sell to Investor the voting common shares of Old Lorus to be received under
the
Arrangement at the same price per share as was paid to shareholders who are
residents of the United States. The proceeds received by the
Selling Shareholders were nominal.
Also
as a condition of the Arrangement, the holder of Old Lorus' secured convertible
debenture agreed to vote in favour of the transaction subject to the repurchase
by New Lorus of its outstanding three million common share purchase warrants
at
a purchase price of $252,000 upon closing of the Arrangement.
The
Company and its subsidiaries have agreed to indemnify Old Lorus and its
directors, officers and employees from and against all damages, losses, expenses
(including fines and penalties), other third party costs and legal expenses,
to
which any of them may be subject arising out of any matter occurring (i)
prior
to, at or after the effective time of the Arrangement (the "Effective Time")
and
directly or indirectly relating to any of the assets of Old Lorus transferred
to
the Company pursuant to the Arrangement (including losses for income, sales,
excise and other taxes arising in connection with the transfer of any such
asset) or conduct of the business prior to the Effective Time; (ii) prior
to, at
or after the Effective Time as a result of any and all interests, rights,
liabilities and other matters relating to the assets transferred by Old Lorus
to
the Company pursuant to the Arrangement; and (iii) prior to or at the Effective
Time and directly or indirectly relating to, with certain exceptions, any
of the
activities of Old Lorus or the Arrangement.
In
connection with the Arrangement the Company and Investor entered into an
escrow
agreement in which $600,000 of the purchase price payable by Investor to
the
Company under was withheld by Investor and placed into escrow with Equity
Transfer & Trust Company, as escrow agent. The monies placed into
escrow will be held as security for and a partial, but not exclusive, source
of
satisfaction of Company’s indemnification obligations to the Investor until the
first anniversary of the Closing Date.
Following
the Arrangement, New Lorus and its subsidiaries have approximately $7.0 million
of unrecognized future tax benefits resulting from non-capital losses carried
forward, and scientific research and experimental development
expenditures. In light of the uncertainty regarding the Company's
ability to generate taxable income in the future, management is of the opinion
that it is more likely than not that these future tax assets will not be
realized in the foreseeable future and hence, a full valuation allowance
will be
recorded against these future tax assets.
-
23
-
Intellectual
Property and Protection of Confidential Information and
Technology
We
believe that our issued patents and pending applications are important in
establishing and maintaining a competitive position with respect to our products
and technology. As of May 31, 2007, we owned or had rights to 46
issued patents and 60 pending patents worldwide.
Antisense-DNA/RNA-based
Theapeutics
We
have been issued two patents in Canada, seven patents in the United States
and
thirteen patents in other jurisdictions around the world relating to our
antisense platform, which include composition of matter and method
claims.
Small
Molecule
We
have been issued two patents in the United States and one patent in Israel
which
include composition of matter and method claims, relating to the NuChem small
molecule platform.
Immunotherapy
We
have been issued two patents in Canada, three patents in the United States
and
10 patents in other jurisdictions around the world relating to our immunotherapy
platform, which include composition of matter, method and process
claims.
Risks
Relating to Intellectual Property
We
either own these issued patents or have the exclusive right to make, use,
market, sell or otherwise commercialize products using these patents to diagnose
and treat cancer. We cannot assure you that we will continue to have exclusive
rights to these patents.
We
cannot assure you that pending applications will result in issued patents,
or
that issued patents will be held valid and enforceable if challenged, or
that a
competitor will not be able to circumvent any such issued patents by adoption
of
a competitive, though non-infringing product or
process. Interpretation and evaluation of pharmaceutical or
biotechnology patent claims present complex and often novel legal and factual
questions. Our business could be adversely affected by increased
competition in the event that any patent granted to it is held to be invalid
or
unenforceable or is inadequate in scope to protect our operations.
While
we believe that our products and technology do not infringe proprietary rights
of others, we cannot assure you that third parties will not assert infringement
claims in the future or that such claims will not be
successful. Furthermore, we could incur substantial costs in
defending ourselves against patent infringement claims brought by others
or in
prosecuting suits against others.
In
addition, we cannot assure you that others will not obtain patents that we
would
need to license, or that if a license is required that it would be available
to
us on reasonable terms, or that if a license is not obtained that we would
be
able to circumvent, through a reasonable investment of time and expense,
such
outside patents. Whether we obtain a license would depend on the
terms offered, the degree of risk of infringement, the vulnerability of the
patent to invalidation and the ease of circumventing the patent.
Until
such time, if ever, that further patents are issued to us, we will rely upon
the
law of trade secrets to the extent possible given the publication requirements
under international patent treaty laws and/or requirements under foreign
patent
laws to protect our technology and our products incorporating the
technology. In this regard, we have adopted certain confidentiality
procedures. These include: limiting access to
confidential
-
24
-
information
to certain key personnel; requiring all directors, officers, employees and
consultants and others who may have access to our intellectual property to
enter
into confidentiality agreements which prohibit the use of or disclosure of
confidential information to third parties; and implementing physical security
measures designed to restrict access to such confidential information and
products. Our ability to maintain the confidentiality of our technology is
crucial to our ultimate possible commercial success. We cannot assure
you that the procedures adopted by us to protect the confidentiality of our
technology will be effective, that third parties will not gain access to
our
trade secrets or disclose the technology, or that we can meaningfully protect
our rights to our technology. Further, by seeking the aforementioned
patent protection in various countries, it is inevitable that a substantial
portion of our technology will become available to our competitors, through
publication of such patent applications.
Regulatory
Strategy
Our
overall regulatory strategy is to work with HC in Canada, the FDA in the
United
States, the EMEA in Europe, and any other local regulatory agencies to have
drug
applications approved for the use of GTI-2040, GTI-2501 and small molecules
in
clinical trials (alone and/or in combination with chemotherapeutic compounds)
and subsequently for sale in international markets. Where possible, we intend
to
take advantage of opportunities for accelerated consideration of drugs designed
to treat rare and serious or life-threatening diseases. We also intend to
pursue
priority evaluation of any application for marketing approval filed in Canada,
the United States or the European Union and to file additional drug applications
in other markets where commercial opportunities exist. We cannot
assure you that we will be able to pursue these opportunities
successfully.
Competition
The
biotechnology and pharmaceutical industries are characterized by rapidly
evolving technology and intense competition. There are many companies
in both these industries that are focusing their efforts on activities similar
to ours. Some of these are companies with established positions in
the pharmaceutical industry and may have substantially more financial and
technical resources, more extensive research and development capabilities,
and
greater marketing, distribution, production and human resources than
us. In addition, we may face competition from other companies for
opportunities to enter into collaborative agreements with biotechnology and
pharmaceutical companies and academic institutions. Many of these
other companies are not solely focused on cancer, as is the mission of our
drug
development. We specialize in the development of drugs that we
believe will manage cancer.
Products
that may compete with our products include chemotherapeutic agents, monoclonal
antibodies, antisense therapies and immunotherapies with novel mechanisms
of
action. These are drugs that are delivered by specific means and are
targeting cancers with large disease populations. We also expect that
we may experience competition from established and emerging pharmaceutical
and
biotechnology companies that have other forms of treatment for the cancers
that
we target. There are many drugs currently in development for the
treatment of cancer that employ a number of novel approaches for attacking
these
cancers. Cancer is a complex disease with more than 100 indications
requiring drugs for treatment. The drugs in competition with our
drugs have specific targets for attacking the disease, targets which are
not
necessarily the same as ours. These competitive drugs therefore could
potentially also be used together in combination therapies with our drugs
to
manage the disease.
Human
Resources
As
at May 31, 2007, we employed 27 full-time persons and three part-time person
in
research and drug development and administration activities. Of our employees,
eight hold Ph.D.s. To encourage a focus on achieving long-term
performance, employees and members of the board of directors have the ability
to
-
25
-
acquire
an ownership interest in the Company through Lorus’ stock option plan and
employees can participate in the employee share purchase plan, which was
established in 2005.
Our
ability to develop commercial products and to establish and maintain our
competitive position in light of technological developments will depend,
in
part, on our ability to attract and retain qualified personnel. There is
a
significant level of competition in the marketplace for such personnel. We
believe that to date we have been successful in attracting and retaining
the
highly skilled personnel critical to our business. We have also chosen to
outsource activities where skills are in short supply or where it is
economically prudent to do so.
None
of our employees are unionized, and we consider our relations with our employees
to be good.
Properties
Our
head office, which occupies 20,500 square feet, is located at 2 Meridian
Road,
Toronto, Ontario. The leased premises include approximately 8,000
square feet of laboratory and research space. We believe that our
existing facilities are adequate to meet our requirements for the near
term. Our current lease expires on March 31, 2008.
Control
of the Registrant
As
of August 29, 2007, to the knowledge of Lorus’ directors and executive officers,
no single Person beneficially owns, directly or indirectly, or exercises
control
or direction over more than 10% of the voting rights attached to all the
outstanding common shares, other than High Tech that held, according to public
filings dated at July 10, 2007 approximately 14% of the issued and outstanding
shares of the company.
RISK
FACTORS
Before
making an investment decision with respect to our common shares, you should
carefully consider the following risk factors, in addition to the other
information included or incorporated by reference into this annual information
form. The risks set out below are not the only risks we face. If any of the
following risks occur, our business, financial condition, prospects or results
of operations would likely suffer. In that case, the trading price of our
common
shares could decline and you may lose all or part of the money you paid to
buy
our common shares.
We
have a history of operating losses. We expect to incur net losses and we
may
never achieve or maintain profitability.
We
have not been profitable since our inception in 1986. We reported net losses
of
$9.6 million; $17.9 million and $22.1 million for the years ended May 31,
2007,
2006 and 2005, respectively. As of May 31, 2007, we had an accumulated deficit
of $174.2 million.
To
date we have only generated nominal revenues from the sale of Virulizin® in
Mexico and we stopped selling Virulizin® in Mexico in July 2005. We have not
generated any other revenue from product sales to date and it is possible
that
we will never have sufficient product sales revenue to achieve profitability.
We
expect to continue to incur losses for at least the next several years as
we or
our collaborators and licensees pursue clinical trials and research and
development efforts. To become profitable, we, either alone or with our
collaborators and licensees, must successfully develop, manufacture and market
our current product candidates, GTI-2040, as well as continue to identify,
develop, manufacture and market new product candidates. It is possible that
we
will never have significant product sales revenue or receive significant
royalties on our licensed product candidates. If funding is insufficient
at any
time in the future, we may not be
-
26
-
able
to develop or commercialize our products, take advantage of business
opportunities or respond to competitive pressures.
Our
current and anticipated operations, particularly our product development
requires substantial capital. We expect that our existing cash and cash
equivalents, along with the funds available to us through the reorganization
agreement described above, will sufficiently fund our current and planned
operations through at least the next twelve months. However, our future capital
needs will depend on many factors, including the extent to which we enter
into
collaboration agreements with respect to any of our proprietary product
candidates, receive royalty and milestone payments from our possible
collaborators and make progress in our internally funded research and
development activities.
Our
capital requirements will also depend on the magnitude and scope of these
activities, our ability to maintain existing and establish new collaborations,
the terms of those collaborations, the success of our collaborators in
developing and marketing products under their respective collaborations with
us,
the success of our contract manufacturers in producing clinical and commercial
supplies of our product candidates on a timely basis and in sufficient
quantities to meet our requirements, competing technological and market
developments, the time and cost of obtaining regulatory approvals, the extent
to
which we choose to commercialize our future products through our own sales
and
marketing capabilities, the cost of preparing, filing, prosecuting, maintaining
and enforcing patent and other rights and our success in acquiring and
integrating complementary products, technologies or companies. We do not
have
committed external sources of funding and we cannot assure you that we will
be
able to obtain additional funds on acceptable terms, if at all. If adequate
funds are not available, we may be required to:
|
|
•
|
engage
in equity financings that would be dilutive to current
shareholders;
|
|
|
•
|
delay,
reduce the scope of or eliminate one or more of our development
programs;
or
|
|
|
•
|
obtain
funds through arrangements with collaborators or others that may
require
us to relinquish rights to technologies, product candidates or
products
that we would otherwise seek to develop or commercialize ourselves;
or
license rights to technologies, product candidates or products
on terms
that are less favourable to us than might otherwise be
available.
|
Our
cash flow may not be sufficient to cover interest payments on our secured
convertible debentures or to repay the debentures at
maturity.
Our
ability to make interest payments, if required to be paid in cash, and to
repay
at maturity or refinance our prime plus 1% convertible debentures due in
approximately 14 months (October 2009) will depend on our ability to generate
or
raise sufficient cash or refinance them. We have never generated positive
annual
cash flow from our operating activities, and we may not generate or sustain
positive cash flows from operations in the future. Our ability to generate
sufficient cash flow will depend on our ability, or the ability of our strategic
partners, to successfully develop and obtain regulatory approval for new
products and to successfully market these products, as well as the results
of
our research and development efforts and other factors, including general
economic, financial, competitive, legislative and regulatory conditions,
many of
which are outside of our control.
We
may violate one or more of the operational covenants related to our convertible
debentures that could result in an event of default and the requirement for
early payment of our convertible debentures.
Our
convertible debentures are subject to certain operational
covenants. In the event that one of those covenants is breached by
us, an event of default could be declared requiring the immediate payment
of the
face value of the debentures. This could result in our inability to
pay and insolvency of the Company, a
-
27
-
dilutive
equity financing in attempt to raise funds to repay the debentures, or a
significant reduction in cash available for us to use towards the development
of
our product candidates.
We
may be unable to obtain partnerships for one or more of our product candidates
which could curtail future development and negatively impact our share
price.
Our
product candidates require significant funding to reach regulatory approval
upon
positive clinical results. Such funding, in particular for
Virulizin®, will be very difficult, or impossible to raise in the public
markets. If such partnerships are not attainable, the development of
these product candidates maybe significantly delayed or stopped
altogether. The announcement of such delay or discontinuation of
development may have a negative impact on our share price.
In
addition, our strategy for the research, development and commercialization
of
our products requires entering into various arrangements with corporate
collaborators, licensers, licensees and others, and our commercial success
is
dependent upon these outside parties performing their respective contractual
responsibilities. The amount and timing of resources that such third-parties
will devote to these activities may not be within our control. We cannot
assure
you that such parties will perform their obligations as expected. We also
cannot
assure you that our collaborators will devote adequate resources to our
programs. In addition, we could become involved in disputes with our
collaborators, which could result in a delay or termination of the related
development programs or result in litigation. We intend to seek additional
collaborative arrangements to develop and commercialize some of our products.
We
may not be able to negotiate collaborative arrangements on favourable terms,
or
at all, in the future, or that our current or future collaborative arrangements
will be successful.
If
we cannot negotiate collaboration, licence or partnering agreements, we may
never achieve profitability.
Clinical
trials are long, expensive and uncertain processes and Health Canada or the
FDA
may ultimately not approve any of our product candidates. We may never develop
any commercial drugs or other products that generate
revenues.
None
of our products has received regulatory approval for commercial use and sale
in
North America. We cannot market a pharmaceutical product in any jurisdiction
until it has completed thorough preclinical testing and clinical trials in
addition to that jurisdiction’s extensive regulatory approval process. In
general, significant research and development and clinical studies are required
to demonstrate the safety and effectiveness of our products before we can
submit
any regulatory applications.
Clinical
trials are long, expensive and uncertain processes. Clinical trials may not
be
commenced or completed on schedule, and Health Canada or the FDA may not
ultimately approve our product candidates for commercial sale. Further, even
if
the results of our preclinical studies or clinical trials are initially
positive, it is possible that we will obtain different results in the later
stages of drug development or that results seen in clinical trials will not
continue with longer term treatment. Drugs in late stages of clinical
development may fail to show the desired safety and efficacy traits despite
having progressed through initial clinical testing. For example, positive
results in early Phase I or Phase II clinical trials may not be repeated
in
larger Phase II or Phase III clinical trials. The results of our
Phase III clinical trial of Virulizinâ did not
meet the
primary endpoint of the study despite promising preclinical and early stage
clinical data. All of our potential drug candidates are prone to the
risks of failure inherent in drug development.
Preparing,
submitting and advancing applications for regulatory approval is complex,
expensive and time intensive and entails significant uncertainty. The results
of
our completed preclinical studies and clinical trials may not be indicative
of
future clinical trial results. A commitment of substantial resources to conduct
time-consuming research, preclinical studies and clinical trials will be
required if we are to complete development
-
28
-
of
our products. Clinical trials of our products require that we identify and
enrol
a large number of patients with the illness under investigation. We may not
be
able to enrol a sufficient number of appropriate patients to complete our
clinical trials in a timely manner particularly in smaller indications such
as
Acute Myeloid Leukemia. If we experience difficulty in enrolling a
sufficient number of patients to conduct our clinical trials, we may need
to
delay or terminate ongoing clinical trials and will not accomplish objectives
material to our success that could affect the price of our common shares.
Delays
in planned patient enrolment or lower than anticipated event rates in our
current clinical trials or future clinical trials may result in increased
costs,
program delays, or both.
In
addition, unacceptable toxicities or adverse side effects may occur at any
time
in the course of preclinical studies or human clinical trials or, if any
products are successfully developed and approved for marketing, during
commercial use of any approved products. The appearance of any such unacceptable
toxicities or adverse side effects could interrupt, limit, delay or abort
the
development of any of our product candidates or, if previously approved,
necessitate their withdrawal from the market. Furthermore, disease resistance
or
other unforeseen factors may limit the effectiveness of our potential
products.
The
clinical trials of any of our drug candidates could be unsuccessful, which
would
prevent us from advancing, commercializing or partnering the drug.
Our
failure to develop safe, commercially viable drugs would substantially impair
our ability to generate revenues and sustain our operations and would materially
harm our business and adversely affect our share price. We may never achieve
profitability.
As
a result of intense competition and technological change in the pharmaceutical
industry, the marketplace may not accept our products or product candidates,
and
we may not be able to compete successfully against other companies in our
industry and achieve profitability.
Many
of our competitors have drug products that have already been approved or
are in
development, and operate large, well-funded research and development programs
in
these fields. Many of our competitors have substantially greater financial
and
management resources, stronger intellectual property positions and greater
manufacturing, marketing and sales capabilities, areas in which we have limited
or no experience. In addition, many of our competitors have significantly
greater experience than we do in undertaking preclinical testing and clinical
trials of new or improved pharmaceutical products and obtaining required
regulatory approvals. Consequently, our competitors may obtain Health Canada,
FDA and other regulatory approvals for product candidates sooner and may
be more
successful in manufacturing and marketing their products than we or our
collaborators are. Existing and future products, therapies and technological
approaches will compete directly with the products we seek to develop. Current
and prospective competing products may provide greater therapeutic benefits
for
a specific problem or may offer easier delivery or comparable performance
at a
lower cost. Any product candidate that we develop and that obtains regulatory
approval must then compete for market acceptance and market share. Our product
candidates may not gain market acceptance among physicians, patients, healthcare
payers and the medical community. Further, any products we develop may become
obsolete before we recover any expenses we incurred in connection with the
development of these products. As a result, we may never achieve
profitability.
If
we fail to attract and retain key employees, the development and
commercialization of our products may be adversely
affected.
We
depend heavily on the principal members of our scientific and management
staff.
If we lose any of these persons, our ability to develop products and become
profitable could suffer. The risk of being unable to retain key personnel
may be
increased by the fact that we have not executed long term employment contracts
with our employees, except for our senior executives. Our future success
will
also depend in large part on our
-
29
-
ability
to attract and retain other highly qualified scientific and management
personnel. We face competition for personnel from other companies, academic
institutions, government entities and other organizations.
We
may be unable to obtain patents to protect our technologies from other companies
with competitive products, and patents of other companies could prevent us
from
manufacturing, developing or marketing our products.
Patent
protection:
The
patent positions of pharmaceutical and biotechnology companies are uncertain
and
involve complex legal and factual questions. The United States (U.S.) Patent
and
Trademark Office and many other patent offices in the world have not established
a consistent policy regarding the breadth of claims that it will allow in
biotechnology patents. Further, allowable patentable subject matter
and the scope of patent protection obtainable may differ between
jurisdictions. If a patent office allows broad claims, the number and
cost of patent interference proceedings in the U.S. or analogous proceedings
in
other jurisdictions and the risk of infringement litigation may increase.
If it
allows narrow claims, the risk of infringement may decrease, but the value
of
our rights under our patents, licenses and patent applications may also
decrease. In addition, the scope of the claims in a patent application can
be
significantly modified during prosecution before the patent is issued.
Consequently, we cannot know whether our pending applications will result
in the
issuance of patents or, if any patents are issued, whether they will provide
us
with significant proprietary protection or will be circumvented, invalidated
or
found to be unenforceable. Until recently, patent applications in the U.S.
were
maintained in secrecy until the patents issued, and publication of discoveries
in scientific or patent literature often lags behind actual discoveries.
Patent
applications filed in the United States after November 2000 generally will
be
published 18 months after the filing date unless the applicant certifies
that
the invention will not be the subject of a foreign patent
application. In many other jurisdictions, such as Canada, patent
applications are published 18 months from the priority date. We
cannot assure you that, even if published, we will be aware of all such
literature. Accordingly, we cannot be certain that the named inventors of
our
products and processes were the first to invent that product or process or
that
we were the first to pursue patent coverage for our inventions.
Enforcement
of intellectual property rights:
Our
commercial success depends in part on our ability to maintain and enforce
our
proprietary rights. If third-parties engage in activities that infringe our
proprietary rights, our management’s focus will be diverted and we may incur
significant costs in asserting our rights. We may not be successful in asserting
our proprietary rights, which could result in our patents being held invalid
or
a court holding that the third-party is not infringing, either of which would
harm our competitive position. In addition, we cannot assure you that others
will not design around our patented technology. Moreover, we may have to
participate in interference proceedings declared by the U.S. Patent and
Trademark Office, European opposition proceedings, or other analogous
proceedings in other parts of the world to determine priority of invention
and
the validity of patent rights granted or applied for, which could result
in
substantial cost and delay, even if the eventual outcome is favourable to
us. We
cannot assure you that our pending patent applications, if issued, would
be held
valid or enforceable. Additionally, many of our foreign patent applications
have
been published as part of the patent prosecution process in such
countries.
Trademark
protection:
Protection
of the rights revealed in published patent applications can be complex, costly
and uncertain. In order to protect goodwill associated with our company and
product names, we rely on trademark protection for our marks. For example,
we
have registered the Virulizin® trademark with the U.S. Patent and Trademark
Office. A third-party may assert a claim that the Virulizin® mark is confusingly
similar to its mark and such claims or the failure to timely register the
Virulizin® mark or objections by the FDA could force us to select a new name for
Virulizin®, which could cause us to incur additional expense.
-
30
-
Trade
secrets:
We
also rely on trade secrets, know-how and confidentiality provisions in our
agreements with our collaborators, employees and consultants to protect our
intellectual property. However, these and other parties may not comply with
the
terms of their agreements with us, and we might be unable to adequately enforce
our rights against these people or obtain adequate compensation for the damages
caused by their unauthorized disclosure or use. Our trade secrets or those
of
our collaborators may become known or may be independently discovered by
others.
Our
products and product candidates may infringe the intellectual property rights
of
others, which could increase our costs.
Our
success also depends on avoiding infringement of the proprietary technologies
of
others. In particular, there may be certain issued patents and patent
applications claiming subject matter which we or our collaborators may be
required to license in order to research, develop or commercialize at least
some
of our product candidates, including Virulizin®, GTI-2040, GTI-2501 and small
molecules. In addition, third-parties may assert infringement or other
intellectual property claims against us based on our patents or other
intellectual property rights. An adverse outcome in these proceedings could
subject us to significant liabilities to third-parties, require disputed
rights
to be licensed from third-parties or require us to cease or modify our use
of
the technology. If we are required to license such technology, we cannot
assure
you that a license under such patents and patent applications will be available
on acceptable terms or at all. Further, we may incur substantial costs defending
ourselves in lawsuits against charges of patent infringement or other unlawful
use of another’s proprietary technology.
If
product liability claims are brought against us or we are unable to obtain
or
maintain product liability insurance, we may incur substantial liabilities
that
could reduce our financial resources.
The
clinical testing and commercial use of pharmaceutical products involves
significant exposure to product liability claims. We have obtained limited
product liability insurance coverage for our clinical trials on humans; however,
our insurance coverage may be insufficient to protect us against all product
liability damages. Further, liability insurance coverage is becoming
increasingly expensive and we might not be able to obtain or maintain product
liability insurance in the future on acceptable terms or in sufficient amounts
to protect us against product liability damages. Regardless of merit or eventual
outcome, liability claims may result in decreased demand for a future product,
injury to reputation, withdrawal of clinical trial volunteers, loss of revenue,
costs of litigation, distraction of management and substantial monetary awards
to plaintiffs. Additionally, if we are required to pay a product liability
claim, we may not have sufficient financial resources to complete development
or
commercialization of any of our product candidates and our business and results
of operations will be adversely affected.
We
have no manufacturing capabilities. We depend on third-parties, including
a
number of sole suppliers, for manufacturing and storage of our product
candidates used in our clinical trials. Product introductions may be delayed
or
suspended if the manufacture of our products is interrupted or
discontinued.
We
do not have manufacturing facilities to produce supplies of Virulizin®,
GTI-2040, GTI-2501, small molecule or any of our other product candidates
to
support clinical trials or commercial launch of these products, if they are
approved. We are dependent on third-parties for manufacturing and storage
of our
product candidates. If we are unable to contract for a sufficient supply
of our
product candidates on acceptable terms, or if we encounter delays or
difficulties in the manufacturing process or our relationships with our
manufacturers, we may not have sufficient product to conduct or complete
our
clinical trials or support preparations for the commercial launch of our
product
candidates, if approved.
Dependence
on contract manufacturers for commercial production involves a number of
risks,
many of which are outside our control. These risks include potential delays
in
transferring technology, and the inability of our
-
31
-
contract
manufacturer to scale production on a timely basis, to manufacture commercial
quantities at reasonable costs, to comply with cGMP and to implement procedures
that result in the production of drugs that meet our specifications and
regulatory requirements.
Our
reliance on contract manufacturers exposes us to additional risks,
including
there
may be delays in scale-up to quantities needed for clinical trials and
commercial launch or failure to manufacture such quantities to our
specifications, or to deliver such quantities on the dates we
require;
our
current and future manufacturers are subject to ongoing, periodic, unannounced
inspection by the FDA and corresponding Canadian and international regulatory
authorities for compliance with strictly enforced cGMP regulations and similar
standards, and we do not have control over our contract manufacturers’
compliance with these regulations and standards;
our
current and future manufacturers may not be able to comply with applicable
regulatory requirements, which would prohibit them from manufacturing products
for us;
if
we need to change to other commercial manufacturing contractors, the FDA
and
comparable foreign regulators must approve these contractors prior to our
use,
which would require new testing and compliance inspections, and the new
manufacturers would have to be educated in, or themselves develop substantially
equivalent processes necessary for the production or our products;
and
our
manufacturers might not be able to fulfill our commercial needs, which would
require us to seek new manufacturing arrangements and may result in substantial
delays in meeting market demand.
Any
of these factors could cause us to delay or suspend clinical trials, regulatory
submission, required approvals or commercialization of our products under
development, entail higher costs and result in our being unable to effectively
commercialize our products. We do not currently intend to manufacture any
of our
product candidates, although we may choose to do so in the future. If we
decide
to manufacture our products, we would be subject to the regulatory risks
and
requirements described above. We would also be subject to similar risks
regarding delays or difficulties encountered in manufacturing our pharmaceutical
products and we would require additional facilities and substantial additional
capital. We cannot assure you that we would be able to manufacture any of
our
products successfully in accordance with regulatory requirements and in a
cost
effective manner.
Our
operations involve hazardous materials and we must comply with environmental
laws and regulations, which can he expensive and restrict how we do
business.
Our
research and development activities involve the controlled use of hazardous
materials, radioactive compounds and other potentially dangerous chemicals
and
biological agents. Although we believe our safety procedures for these materials
comply with governmental standards, we cannot entirely eliminate the risk
of
accidental contamination or injury from these materials. We currently have
insurance, in amounts and on terms typical for companies in businesses that
are
similarly situated, that could coverall or a portion of a damage claim arising
from our use of hazardous and other materials. However, if an accident or
environmental discharge occurs, and we are held liable for any resulting
damages, the associated liability could exceed our insurance coverage and
our
financial resources.
We
have limited sales, marketing and distribution
experience.
We
have very limited experience in the sales, marketing and distribution of
pharmaceutical products. There can be no assurance that we will be able to
establish sales, marketing, and distribution capabilities or make
-
32
-
arrangements
with our collaborators, licensees or others to perform such activities or
that
such efforts will be successful. If we decide to market any of our products
directly, we must either acquire or internally develop a marketing and sales
force with technical expertise and with supporting distribution capabilities.
The acquisition or development of a sales and distribution infrastructure
would
require substantial resources, which may divert the attention of our management
and key personnel and have a negative impact on our product development efforts.
If we contract with third-parties for the sales and marketing of our products,
our revenues will be dependent on the efforts of these third-parties, whose
efforts may not be successful. If we fail to establish successful marketing
and
sales capabilities or to make arrangements with third-parties, our business,
financial condition and results of operations will be materially adversely
affected.
Our
interest income is subject to fluctuations of interest rates in our investment
portfolio.
Our
investments are held to maturity and have staggered maturities to minimize
interest rate risk. There can be no assurance that interest income fluctuations
will not have an adverse impact on our financial condition. We maintain all
our
accounts in Canadian dollars, but a portion of our expenditures are in foreign
currencies. We do not currently engage in hedging our foreign currency
requirements to reduce exchange rate risk.
Because
of the uncertainty of pharmaceutical pricing, reimbursement and healthcare
reform measures, if any of our product candidates are approved for sale to
the
public, we may be unable to sell our products
profitably.
The
availability of reimbursement by governmental and other third-party payers
affects the market for any pharmaceutical product. These third-party payers
continually attempt to contain or reduce the costs of healthcare. There have
been a number of legislative and regulatory proposals to change the healthcare
system and further proposals are likely. Significant uncertainty exists with
respect to the reimbursement status of newly approved healthcare products.
In
addition, third-party payers are increasingly challenging the price and cost
effectiveness of medical products and services. We might not be able to sell
our
products profitably or recoup the value of our investment in product development
if reimbursement is unavailable or limited in scope.
RISKS
RELATED TO OUR COMMON SHARES AND CONVERTIBLE DEBENTURES
Our
share price has been and may continue to be volatile and an investment in
our
common shares could suffer a decline in value.
You
should consider an investment in our common shares as risky and invest only
if
you can withstand a significant loss and wide fluctuations in the market
value
of your investment. We receive only limited attention by securities analysts
and
frequently experience an imbalance between supply and demand for our common
shares. The market price of our common shares has been highly volatile and
is
likely to continue to be volatile. Factors affecting our common share price
include:
|
|
•
|
the
progress of our and our collaborators’ clinical trials, including our and
our collaborators’ ability to produce clinical supplies of our product
candidates on a timely basis and in sufficient quantities to meet
our
clinical trial requirements;
|
|
|
•
|
announcements
of technological innovations or new product candidates by us, our
collaborators or our competitors;
|
|
|
•
|
fluctuations
in our operating results;
|
|
|
•
|
published
reports by securities analysts;
|
-
33
-
|
|
•
|
developments
in patent or other intellectual property
rights;
|
|
|
•
|
publicity
concerning discovery and development activities by our
licensees;
|
|
|
•
|
the
cash and short term investments held us and our ability to secure
future
financing;
|
|
|
•
|
public
concern as to the safety and efficacy of drugs that we and our
competitors
develop;
|
|
|
•
|
governmental
regulation and changes in medical and pharmaceutical product reimbursement
policies; and
|
|
|
•
|
general
market conditions.
|
Future
sales of our common shares by us or by our existing shareholders could cause
our
share price to fall.
Additional
equity financings or other share issuances by us could adversely affect the
market price of our common shares. Sales by existing shareholders of a large
number of shares of our common shares in the public market and the sale of
shares issued in connection with strategic alliances, or the perception that
such additional sales could occur, could cause the market price of our common
shares to drop.
Conversion
of our secured convertible debentures will dilute the ownership interest
of
existing shareholders.
The
conversion of some or all of the convertible debentures will dilute the
ownership interests of existing shareholders. Any sales in the public market
of
the common shares issuable upon such conversion could adversely affect
prevailing market prices of our common shares. In addition, the existence
of the
secured convertible debentures may encourage short selling by market
participants.
DIVIDENDS
Dividends
on our common shares are declared at the discretion of our board of
directors. To date, we have not paid any dividends and do not expect
to do so in the foreseeable future.
SHARE
CAPITAL AND MARKET FOR SECURITIES
Share
Capital
We
are authorized to issue an unlimited number of common shares. As of
August 29, 2007, there were 212,627,876 common shares issued and
outstanding. In addition, as of August 29, 2007 there were 12,494,389
common shares issuable upon the exercise of outstanding stock options at
a
weighted average exercise price of $0.59 per share, the total number of common
shares issuable under the 2003 plan is 31,894,181 of which the company has
applied to the TSX to list 5,592,097 of common shares available for future
issuance under the Company’s equity compensation plans. The
holders of common shares are entitled to one vote per share at meetings of
shareholders, to receive such dividends as declared by us and to receive
our
remaining property and assets upon our dissolution or winding up. Our common
shares are not subject to any future call or assessment and there are no
pre-emptive, conversion or redemption rights attached to such
shares.
-
34
-
Market
for Securities
Our
common shares are currently listed on The Toronto Stock Exchange (“TSX”) under
the symbol “LOR” and on the American Stock Exchange under the symbol
“LRP”. The following table sets out the price ranges and trading
volumes of our common shares on the TSX for the periods indicated:
|
High
($)
|
Low
($)
|
Volume
(#)
|
|||
|
2007
|
|||||
|
May
|
0.29
|
0.25
|
4,667,853
|
||
|
April
|
0.33
|
0.26
|
7,270,442
|
||
|
March
|
0.33
|
0.26
|
3,425,904
|
||
|
February
|
0.39
|
0.28
|
10,487,394
|
||
|
January
|
0.33
|
0.23
|
12,159,851
|
||
|
2006
|
|||||
|
December
|
0.28
|
0.22
|
5,822,876
|
||
|
November
|
0.29
|
0.22
|
3,623,228
|
||
|
October
|
0.30
|
0.23
|
6,378,136
|
||
|
September
|
0.34
|
0.28
|
3,882,530
|
||
|
August
|
0.39
|
0.30
|
4,281,069
|
||
|
July
|
0.38
|
0.28
|
2,150,396
|
||
|
June
|
0.37
|
0.31
|
2,334,732
|
||
Principal
Shareholders
To
our knowledge, based on publicly available information, the only persons
or
entities that own more than 5% of our issued and outstanding common shares
are
Technifund Inc. and its related parties, which currently owns 9.8%
of our issued and outstanding common shares and High Tech
that held,
approximately 14% of the issued and outstanding shares of the
company. See Business of the Company - Financial
Strategy”.
DIRECTORS
AND OFFICERS
The
following table and notes thereto provide the name, province or state and
country of residence, positions with the Company and term of office of each
person who serves as a director or executive officer of Lorus as at the date
hereof.
Each
director has been elected or appointed to serve until the next annual meeting
or
until a successor is elected or appointed. We have an Audit
Committee, an Environmental, Health and Safety Committee, a Corporate Governance
and Nominating Committee and a Compensation Committee the members of each
such
committee are shown below. As at May 31, 2007, our directors and
executive officers, as a group, beneficially owned, directly or indirectly,
or
exercised control over approximately 33,750,000 common shares or approximately
16% of our outstanding common shares.
-
35
-
|
Name
and Province/State and
Country
of Residence
|
Position
|
Director
or Officer Since
|
|
J.
Kevin Buchi(1)
Pennsylvania,
United States
|
Director
|
December
2002
|
|
Donald
W. Paterson(1)(3)
Ontario,
Canada
|
Director
|
July
1991
|
|
Alan
Steigrod(2)
Florida,
United States
|
Director
|
May
2001
|
|
Georg
Ludwig(2)
Eschen,
Liechtenstein
|
Director
|
September
2006
|
|
Michael
Moore(2)
Surrey,
United Kingdom
|
Director
|
September
2006
|
|
Graham
Strachan(1))(3)(4)
Ontario,
Canada
|
Chairman,
Director
|
May
2001
|
|
Dr.
Jim Wright
Ontario,
Canada
|
Director,
Former President and Chief Executive Officer, Director
|
October
1999
|
|
Dr.
Aiping Young(4)
Ontario,
Canada
|
President
and Chief Executive Officer, former Chief Operating
Officer
|
October
1999
|
|
Elizabeth
Williams
Ontario,
Canada
|
Director
of Finance and Acting Chief Financial Officer
|
November
2005
|
(1) Member
of Audit Committee.
(2) Member
of the Compensation Committee.
(3) Member
of the Corporate Governance and Nominating Committee.
(4) Member
of Environment, Health and Safety Committee.
The
principal occupation and employment of each of the foregoing persons for
the
past five years is set forth below:
J.
Kevin Buchi: Mr. Buchi is Executive Vice President and Chief Financial
Officer of Cephalon Inc., an international biopharmaceutical
company. Mr. Buchi is responsible for finance, accounting,
manufacturing and information systems and has been involved in raising
significant financing for Cephalon. He is a certified public accountant and
has
received a master’s degree in management from the J.L. Kellogg Graduate School
of Management at Northwestern University.
Donald
W. Paterson: Mr. Paterson is President of Cavandale Corporation,
a corporation principally engaged in providing strategic corporate consulting
to
emerging growth companies within the technology industry.
Alan
Steigrod: Mr. Steigrod is Managing Director of Newport
Healthcare Ventures, a consulting firm for the healthcare industry, located
in
Newport Beach, California.
-
36
-
Georg
Ludwig: Mr. Ludwig is Managing Director of ConPharm Anstalt a
consulting and managment company for life science funds, located in
Lechteinstein.
Michael
Moore: Mr. Moore is Chief Executive Officer, Piramed Limited a
biopharmaceutical specializing in new classes of small molecule anti-tumour
agents.
Graham
Strachan: Mr. Strachan is President of GLS Business Development
Inc., a life-science consulting firm located in Etobicoke, Ontario.
Dr.
Jim Wright: Dr. Wright is presently Chief Executive Officer
of NuQuest Bio Inc. Dr. Wright co-founded GeneSense Technologies
Inc. in 1996, and served as Lorus' President, Chief Scientific Officer and
a member of the Board of Directors in October 1999 on a merger
with GeneSense. In September 2006 he stepped down as the
President and Chief Executive Officer of Lorus.
Dr.
Aiping Young: Dr. Young has been our President and Chief
Executive Officer since September 21, 2006 and was a cofounder with Dr. Wright
of GeneSense Technologies Inc. Dr. Young previously held the position
of Chief Operating Officer, Senior Vice President, Research and Development
and
Chief Technical Officer at Lorus.
Elizabeth
Williams: Prior to joining Lorus in July 2004, Ms. Williams was an Audit
Manager with Ernst and Young LLP. Ms. Williams is a chartered
accountant and has received a bachelor’s degree in business
administration. Ms. Williams lectured on introductory auditing
at Wilfrid Laurier University during 2005.
COMMITTEE
INFORMATION
The
Audit Committee Charter of Old Lorus was adopted by the Company in connection
with the Arrangement.
Audit
Committee
The
charter of our audit committee is attached as Schedule A. The current
members of the audit committee are J. Kevin Buchi, Donald W. Paterson and
Graham
Strachan. Pursuant to Canadian securities laws, our board of
directors has determined that Messrs. Buchi, Paterson and Strachan are
financially literate as all have experience in reviewing and analysing the
financial reports and ascertaining the financial position of a
corporation. Mr. Buchi is a certified public accountant and holds the
position of Chief Financial Officer in a public pharmaceutical
company. Pursuant to United States securities laws, Mr. Buchi is also
an audit committee “financial expert”. Mr. Paterson, in his position
as President of Cavandale Corporation, is educated and experienced in reading
and analyzing financial statements. Mr. Strachan has experience with
reading and analysing financial statements both as President of his own life
science consulting firm and in a prior position as President, Chief Executive
Officer and a director of a biopharmaceutical company. Additionally,
we believe that all three members of the audit committee qualify as
“independent” as that term is defined in the relevant Canadian and United States
securities laws relating to the composition of the audit committee.
Independent
Auditors
Auditor’s
Fees
The
total fees billed for professional services by KPMG LLP (our independent
auditors) for the years ended May 31, 2007 and 2006 are as follows:
-
37
-
|
2007
|
2006
|
|
|
Audit
Fees
|
$330,000
|
$198,500
|
|
Tax
Fees
|
$8,500
|
$13,100
|
|
Total
|
$338,500
|
$211,600
|
Audit
fees consist of the fees paid with respect to the audit of our consolidated
annual financial statements, quarterly reviews and accounting assistance
and
fees fro services associated with the filing of the management proxy circular
in
May 2007 amounting to $150,000. Tax fees relate to assistance provided with
respect of proposed transactions and review of tax returns.
Pre-Approval
Policies and Procedures
The
audit committee of our board of directors has, pursuant to the audit committee
charter, adopted specific responsibilities and duties regarding the provision
of
services by our external auditors, currently KPMG LLP. Our charter
requires audit committee pre-approval of all permitted audit and audit-related
services. Any non-audit services must be submitted to the audit
committee for review and approval. Under the charter, all permitted
services to be provided by KPMG LLP must be pre-approved by the audit
committee.
Subject
to the charter, the audit committee may establish fee thresholds for a group
of
pre-approved services. The audit committee then recommends to the
board of directors approval of the fees and other significant compensation
to be
paid to the independent auditors.
No
services were provided by KPMG LLP under a de minimus exemption for our
fiscal years ended May 31, 2007 and 2006.
LEGAL
PROCEEDINGS
We
are not a party to, nor the subject of, any outstanding legal proceedings,
nor
are we aware of any contemplated proceedings.
TRANSFER
AGENT AND REGISTRAR
The
transfer agent and registrar for our common shares is Computershare Investor
Services Inc. at its principal office in the City of
Toronto.
MATERIAL
CONTRACTS
Other
than the agreements described below, we have not, during our financial year
ending May 31, 2007, entered into any material agreements other than contracts
in the ordinary course of business. Agreements completed prior to
July 10, 2007 are filed on SEDAR under Old Lorus (4322531 Canada Inc.) and
those
completed after July 10, 2007 are filed on SEDAR under New Lorus.
|
1.
|
Subscription
Agreement dated July 13, 2006 between the Company and HighTech.
See
“Business of the Company - Financial Strategy - Share
Issuances”.
|
|
2.
|
Subscription
Agreement dated July 24, 2006 between the Company and Technifund.
See
“Business of the Company - Financial Strategy - Share
Issuances”.
|
-
38
-
|
3.
|
Registration
Rights Agreement dated August 30, 2006 between the Company and
High Tech
under which certain rights were granted to High Tech, including
the right
to require the Company to file a Canadian prospectus or a United
States
Registration Statement and the right to require the Company to
include in
any public offering such number of securities of the Company held
by High
Tech as High Tech may request..
|
|
4.
|
Arrangement
Agreement dated May 1, 2007 between the Company, Old Lorus, 6707157
Canada
Inc., NuChem Pharmaceuticals Inc. (“NuChem”), Genesense Technologies Inc.
(“Genesense”) and Pinnacle International Lands Inc., as amended May 14,
2007 and July 4, 2007. See “Business of the Company - Financial Strategy -
Plan of Arrangement and Corporate
Reorganization”.
|
|
5.
|
Warrant
Repurchase Agreement dated May 1, 2007 between the Company and
TEMIC. See
“Business of the Company - Financial Strategy - Secured Convertible
Debentures”.
|
|
6.
|
Assignment,
Novation and Amendment Agreement and Consent dated May 1, 2007
among the
Company, Old Lorus, Genesense and TEMIC as amended June 28, 2007
under
which the Company assumed Old Lorus’ obligation to pay TEMIC the $15
million aggregate principal amount of the Debentures plus accrued
unpaid
interest thereon in consideration for Old Lorus issuing a non-interest
bearing promissory note.
|
|
7.
|
Tangible
Business Assets Transfer Agreement dated July 10, 2007 between
Old Lorus
and Genesense under which Old Lorus transferred certain depreciable
property to Genesense, as contemplated in the plan of
arrangement.
|
|
8.
|
Antisense
Patent Transfer Agreement dated July 10, 2007 between the Company
and
Genesense under which Genesense transferred certain Antisense patent
assets to the Company in exchange for a demand non-interest bearing
promissory note issued by the
Company.
|
|
9.
|
Virulizin
and Small Molecule Patent Assets Transfer Agreement dated July
10, 2007
between Old Lorus and Genesense under which Old Lorus transferred
Virulizin and Small Molecule Patent Assets to Genesense in consideration
for the issuance by Genesense of one common share of
Genesense.
|
|
10.
|
Prepaid
Expenses and Receivables Transfer Agreement dated July 10, 2007
between
Old Lorus and Genesense under which Old Lorus transferred certain
prepaid
expenses and receivables to Genesense in exchange for the issuance
by
Genesense of one common share of
Genesense.
|
|
11.
|
Share
Purchase Agreement dated July 10, 2007 under which Old Lorus transferred
all of the common shares of NuChem held by it to the Company at
a price
equal to their fair market value in consideration for the issuance
of a
demand non-interest bearing promissory
note.
|
|
12.
|
Share
Purchase Agreement dated July 10, 2007 under which Old Lorus transferred
all of the common shares of Genesense held by it to the Company
at a price
equal to their fair market value in exchange for the assumption
by the
Company of Old Lorus’ remaining liabilities and the issuance of a demand
non-interest bearing promissory
note.
|
|
13.
|
Share
purchase agreement dated July 10, 2007 under which the Company
transferred
certain shares of Old Lorus held by it to 6707157 Canada Inc. in
consideration of a cash payment as specified in the plan of arrangement,
subject to payment and adjustment in accordance with such agreement
and a
holdback to an escrow agreement.
|
-
39
-
|
14.
|
Indemnification
Agreement dated July 10, 2007 between Old Lorus and the Company.
See
“Business of the Company - Financial Strategy - Plan of Arrangement
and
Corporate Reorganization”.
|
|
15.
|
Escrow
Agreement between 6707157 Canada Inc, the Company and Equity Transfer
& Trust Company dated July 10, 2007 providing for an escrow amount
related to the plan of arrangement. See “Business of the Company -
Financial Strategy - Plan of Arrangement and Corporate
Reorganization”.
|
|
16.
|
Amended
and Restated Guarantee and Indemnity between GeneSense and TEMIC
dated
July 10, 2007 reaffirming TEMIC’s guaranties and indemnities in respect of
TEMIC’s Debentures.
|
|
17.
|
Amended
and Restated Share Pledge Agreement between the Company and TEMIC
dated
July 10, 2007 reaffirming the Company’s pledge of shares in its
subsidiaries in respect of TEMIC’s
Debentures.
|
INTERESTS
OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
None
of our directors, executive officers or to our knowledge, principal
shareholders, or any associate or affiliate of the forgoing, has had any
material interest, direct or indirect, in any transaction within the three
most
recently completed financial years or during the current financial year prior
to
the date of this annual information form that has materially affected or
will
materially affect us.
INTERESTS
OF EXPERTS
KPMG
LLP, the Company’s external auditor, has reported on the consolidated financial
statements of the Company for each of the years in the three-year period
ended
May 31, 2006. KPMG LLP is independent of Lorus in accordance with the
applicable Rules of Professional Conduct/Code of Ethics of the Institute
of
Chartered Accountants of Ontario, and within the meaning of the Securities
Acts
administered by the United States Securities and Exchange Commission and
the
requirements of the Independence Standards Board.
ADDITIONAL
INFORMATION
Additional
information relating to Lorus may be found on SEDAR at
www.sedar.com. Certain additional information, including directors’
and officers’ remuneration and indebtedness, principal holders of our
securities, and securities authorized for issuance under our stock option
plan,
is contained in the Company’s management information circular dated August 15,
2007 for the September 19, 2007 annual meeting of shareholders (the
“Circular”). Additional financial information is provided in our
financial statements and management’s discussion and analysis for the financial
year ended May 31, 2007 (the “2007 Financial Statements”). Copies
of:
|
|
•
|
the
Circular;
|
|
|
•
|
the
2007 Financial Statements and our most recent unaudited financial
statements that have been filed, if any, for any period subsequent
to the
year ended May 31, 2007;
|
|
|
•
|
this
annual information form and any document or the pertinent pages
of any
document incorporated by reference in this annual information form;
and
|
|
|
•
|
when
our securities are in the course of a distribution pursuant to
a short
form prospectus or a preliminary short form prospectus, one copy
of any
other documents that are incorporated by reference into the short
form
prospectus or preliminary short form prospectus otherwise not referred
to
herein,
|
-
40
-
may
be obtained upon request from our Director of Finance at our offices located
at
2 Meridian Road, Toronto, Ontario, M9W 4Z7, Canada. If our securities
are in the course of a distribution pursuant to a short form prospectus or
a
preliminary short form prospectus, copies of the foregoing documents are
available free of charge. At all other times, a reasonable fee may be
charged if a person who is not a security holder of Lorus makes the request
for
copies.
-
41
-
GLOSSARY
The
following is a glossary of terms that are used in this annual information
form:
|
Analog:
|
a
chemical derivative or variation of a parent molecule
|
|
Anti-proliferative:
|
preventing
cell division
|
|
Apoptosis:
|
programmed
cell death
|
|
Carcinoma:
|
any
cancerous tumor that starts with the cells that cover the inner
and outer
body surfaces
|
|
Clinical
trials:
|
the
investigational use of a new drug in humans: Phase I clinical trials
test a drug for safety, Phase II clinical further test for safety and
may test for efficacy in a relatively small sample of patients
and
Phase III clinical trials test the drug for efficacy in larger
numbers of patients and compares the drug with conventional
therapies
|
|
cGMP:
|
current
good manufacturing practices, as mandated from time to time by
the HC and
the FDA and EMEA
|
|
Complete
response:
|
When
all signs of cancer disappear in response to treatment. This is
based on symptoms, physical exam, and radiology and lab
tests. This does not always mean the cancer has been cured.
Also called complete remission.
|
|
CLT:
|
clotrimazole
|
|
Cytokine:
|
a
generic term for a non-antibody protein released by a cell population
(e.g., activated macrophages) of the immune system on contact with
chemical or biological stimuli
|
|
Cytotoxic:
|
pertaining
to the destruction of cells
|
|
Deoxyribonucleic
acid (DNA):
|
DNA
is the carrier of genetic information which exists in all cells
of the
body. The building blocks of DNA are called
nucleotides
|
|
Disease
stabilization:
|
“no
change” category for clinical response, that is, no increase or decrease
in tumour dimensions or change in extent or severity of disease
state as
pre-defined in a clinical protocol. Usually requires more than
one
measurement of stable disease and/or stable disease over a pre-determined
length of time
|
|
ECOG:
|
Eastern
Cooperative Oncology Group
|
|
Efficacy:
|
the
ability of a drug to produce a desired result
|
|
Efficacy
evaluable population:
|
patients
that meet pre-defined protocol requirements (criteria usually found
in the
Statistical Analysis Plan) for inclusion in efficacy evaluation
datasets.
|
|
EMEA:
|
European
Medicine Evaluation Agency
|
-
42
-
|
FDA:
|
Food
and Drug Administration, the government agency which regulates
the use and
sale of diagnostic and therapeutic drug products in the United
States
|
|
Gene
expression:
|
the
synthesis of specific proteins on the basis of inherited or acquired
genetic information
|
|
GeneSense:
|
GeneSense
Technologies Inc.
|
|
HC:
|
Health
Canada, the federal government department which among other
responsibilities regulates the use and sale of therapeutic drug
products
in Canada
|
|
Immune
system:
|
the
totality of organs and cells involved in the body’s immunologic response
to foreign antigens and malignant tissue
|
|
IND:
|
investigational
new drug
|
|
In
vitro:
|
in
the test tube; referring to chemical reactions, fermentation, etc.,
occurring therein e.g., in cell-free extracts
|
|
In
vivo:
|
in
the living body; referring to chemical processes occurring within
cells,
etc., as distinguished from those occurring in cell-free extracts
(in vitro)
|
|
Krüppel-like
factor 4:
|
an
epithelial cell-enriched, zinc finger-containing transcription
factor, the
expression of which is associated with growth arrest
|
|
Macrophage:
|
a
large scavenger white blood cell that engulfs and digests invading
micro-organisms and cell debris, and also participates in many
complex
immunologic processes
|
|
Malignant/
malignancy:
|
describes
a tumor that is cancerous. Two important qualities of
malignancies are the tendency to invade surrounding tissues and
to break
off and spread elsewhere (metastasis)
|
|
MAP
Kinase Pathway:
|
the
pathway of mitogenic signal transduction through the cascade of
mitogen-activated protein (MAP) kinases which ultimately lead to
alteration in regulatory events such as cell proliferation,
differentiation and apoptosis.
|
|
Metabolism:
|
the
overall biochemical reactions that take place in a living organism
including the building up of complex molecules or breakdown of
molecules
to provide energy
|
|
Metabolic:
|
of,
or relating to, the metabolism
|
|
Metastasis:
|
the
process by which tumor cells are spread to other parts of the
body
|
|
Monocyte:
|
a
large white blood cell with finely granulated chromatin dispersed
throughout the nucleus that is formed in the bone marrow, enters
the
blood, and migrates into the connective tissue where it differentiates
into a macrophage
|
-
43
-
|
mRNA:
|
messenger,
or mRNA, is a copy of the information carried by a gene on the
DNA. The role of mRNA is to move the information contained in
DNA to the translation machinery.
|
|
NDA:
|
new
drug application, the application to obtain marketing approval
filed with
the FDA or BCD after completion of human clinical
trials
|
|
NDS:
|
new
drug submission, the application to obtain marketing approval filed
with
the HC after completion of human clinical trials
|
|
NOC:
|
Notice
of Compliance
|
|
NuChem:
|
NuChem
Pharmaceuticals Inc.
|
|
NuChem
Analogs:
|
analogs
of CLT licensed by us for anticancer indications
|
|
Nucleic
acid:
|
DNA
and RNA, each of which are formed by the combination of nucleotides;
it is
found in all living cells and contains the genetic code required
to
transfer genetic information from one generation to the
next
|
|
Nucleotide:
|
a
compound consisting of a purine or pyrimidine base, a pentose sugar
and a
phosphoric acid; they are the building blocks from which nucleic
acids
(DNA or RNA) are constructed
|
|
Pharmacokinetics:
|
the
action of drugs in the body over a period of time, including the
process
of absorption, distribution, localization in tissues, biotransformation
and excretion
|
|
Pre-clinical
testing:
|
testing
that is conducted in the laboratory (chemistry and pharmacology)
and with
animals to help determine a product’s chemical, pharmacological and
pharmaceutical characteristics (including mechanism of action),
toxicity,
efficacy and side effects
|
|
Proteins:
|
large
molecules composed of long chains of sub-units of amino
acids
|
|
PSA
response:
|
a
measured decrease in the levels of prostate specific antigen in
patients
receiving treatment for prostate cancer. Clinically significant
response
defined within a clinical protocol, i.e. 50% reduction in PSA levels
measured at least twice over a defined period of time. PSA is a
substance
produced by the prostate that may be found in elevated amounts
in the
blood of men who have prostate cancer or other medical conditions
affecting the prostate
|
|
R1
and R2:
|
components
of ribonucleotide reductase
|
|
Ribonucleic
acid (RNA):
|
a
nucleic acid found in both the nucleus and the cytoplasm of all
cells. It carries genetic information from the nucleus to the
cytoplasm, where it also reacts as a template in association with
ribosomes to synthesize
proteins
|
-
44
-
|
Single-arm
pilot study:
|
a
pilot study is usually an initial study examining a new method
or
treatment. A single-arm clinical study is when a drug is administered
to a
single group of patients and the results are compared to historical
data
of untreated patients. These studies do not have a control arm
and
typically enrol a small number of patients.
|
|
siRNA:
|
a
short sequence of RNA that can decrease gene expression in a highly
specific manner (gene silencing).
|
|
Toxicity:
|
a
condition that results from exposure to a substance at levels causing
deleterious side effects which may be harmful to an
organism
|
|
Tumor:
|
an
abnormal swelling or lump in the body caused by the growth of new
tissues
which differ in structure from the part of the body in which they
are
growing. A tumor may be benign or malignant
|
|
Tumor
necrosis:
|
tumor
deterioration and death
|
|
Xenograft:
|
an
implant of a foreign substance
|
-
45
-
SCHEDULE
A
CHARTER
OF THE AUDIT COMMITTEE OF THE BOARD OF DIRECTORS
OF
LORUS THERAPEUTICS INC. (the “Company”)
|
18.
|
PURPOSE
|
The
Audit Committee is a committee of the board of directors of the Company (the
“Board”). The primary function of the Audit Committee is to assist the Board in
fulfilling its oversight responsibilities. The Audit Committee’s primary duties
and responsibilities are to:
|
|
(a)
|
serve
as an independent and objective party to monitor the integrity
of the
Company’s financial reporting process and systems of internal controls
regarding finance, accounting, and legal
compliance;
|
|
|
(b)
|
identify
and monitor the management of the principal risks that could impact
the
financial reporting of the Company;
|
|
|
(c)
|
monitor
the independence and performance of the Company’s independent
auditors;
|
|
|
(d)
|
provide
an avenue of communication among the independent auditors, management,
and
the Board; and
|
|
|
(e)
|
encourage
continuous improvement of, and foster adherence to, the Company’s
policies, procedures and practices at all
levels.
|
The
Audit Committee has the authority to conduct any investigation appropriate
to
fulfilling its responsibilities, and it has direct access to the independent
auditors as well as anyone in the Company. The Audit Committee has the ability
to retain, at the Company’s expense, special legal, accounting, or other
consultants or experts it deems necessary in the performance of its
duties.
|
19.
|
COMPOSITION
AND MEETINGS
|
Audit
Committee members shall meet the requirements of Canadian and United States
securities laws, including the requirements of the stock exchanges on which
the
Company’s securities are listed.
The
Audit Committee shall be comprised of three or more directors as determined
by
the Board, each of whom shall be independent as defined by Multilateral
Instrument 52-110 - Audit Committees of the Canadian Securities Administrators
(“MI 52-110”), United States securities laws, and applicable
stock exchange rules. All members of the Audit Committee shall have a
basic understanding of finance and accounting and be able to read and understand
fundamental financial statements, and at least one member of the Committee
shall
have accounting or related financial management expertise.
Audit
Committee members shall be appointed by the Board. If an Audit Committee
Chair
is not designated or present, the members of the Audit Committee may designate
a
Chair by majority vote of the Audit Committee membership.
The
Audit Committee shall meet at least four times annually, or more frequently
as
circumstances require. The Audit Committee Chair shall prepare and/or approve
an
agenda in advance of each meeting.
-
46
-
The
Audit Committee may ask members of management or others to attend meetings
and
provide pertinent information as necessary. The Audit Committee should meet
privately in executive session at least annually with management, the
independent auditors, and as a committee to discuss any matters that the
Audit
Committee or each of these groups believe should be discussed. In addition,
the
Audit Committee should communicate with management quarterly to review the
Company’s financial statements.
|
20.
|
RESPONSIBILITIES
AND DUTIES
|
|
|
(a)
|
Review
Procedures
|
|
|
(i)
|
Maintain
a Charter that sets out the Audit Committees mandate and
responsibilities. Review and reassess the adequacy of this
Charter at least annually.
|
|
|
(ii)
|
Review
the Company’s financial statements, MD&A and annual and interim
results press releases prior to filing or distribution. The
Audit Committee must be satisfied that adequate procedures are
in place
for the review of the Company’s public disclosure of financial information
extracted or derived from the Company’s financial statements (other than
public disclosure of financial statements, MD&A and annual and interim
results press releases), and must periodically assess the adequacy
of
those procedures. Consider the independent auditors’ judgements
about the quality and appropriateness, not just the acceptability,
of the
Company’s accounting principles and financial disclosure practices, as
applied in its financial reporting, particularly about the degree
of
aggressiveness or conservatism of its accounting principles and
underlying
estimates and whether those principles are common practices or
are
minority practices.
|
|
|
(iii)
|
Consider
and approve, if appropriate, major changes to the Company’s accounting
principles and practices as suggested by the independent auditors
or
management and assure that the reasoning is described in determining
the
appropriateness of changes in accounting principles and
disclosures.
|
|
|
(iv)
|
In
consultation with the management and the independent auditors,
consider
the integrity of the Company’s financial reporting processes and controls.
Discuss significant financial risk exposures and the steps management
has
taken to monitor, control, and report such exposures. Review significant
findings prepared by the independent auditors together with management’s
responses.
|
|
|
(v)
|
The
Audit Committee is directly responsible for the appointment, compensation,
retention and oversight of the work of the independent auditors
including
the review of any disagreements between management and the independent
auditors in connection with the preparation of the financial statements
and overseeing the resolution of any such
disagreements.
|
|
|
(vi)
|
Annually
review policies and procedures as well as audit results associated
with
directors’ and officers’ expense accounts and perquisites. Annually review
a summary of director and officers’ related party transactions and
potential conflicts of interest.
|
|
|
(vii)
|
Annually
conduct self-assessment of Audit Committee performance including
a review
and discussion of the Audit Committee roles and responsibilities,
seeking
input from senior management, the full Board and others if
needed.
|
-
47
-
|
|
(b)
|
Independent
Auditors
|
|
|
(i)
|
The
independent auditors are directly accountable to the Audit Committee
and
the Board, and shall report directly to the Audit Committee. The
Audit
Committee shall review the independence and performance of the
auditors
and annually recommend to the
Board:
|
|
|
A.
|
The
external auditor to be nominated for the purpose of preparing or
issuing
an auditor’s report and performing other audit, review and attest services
for the Company as required;
|
|
|
B.
|
The
compensation of the auditor; and
|
|
|
C.
|
To
approve any discharge of the auditors when circumstances
warrant.
|
|
|
(ii)
|
Pre-approve
all audit fees and terms and all permitted non-audit services provided
by
the external auditor, and consider whether these services are compatible
with the auditors’ independence. Any member of the Audit
Committee may approve additional proposed permitted non-audit services
that arise between Audit Committee meetings provided that the decision
to
pre-approve the services is presented at the next scheduled Audit
Committee meeting. The approval of all non-audit services will
be evidenced by the completion and approval of the Non-Audit Services
Request Form (attached as Appendix “A”
hereto).
|
|
|
(iii)
|
On
an annual basis, the Audit Committee should review and discuss
with the
independent auditors all significant relationships they have with
the
Company that could impair the auditors’
independence.
|
|
|
(iv)
|
Review
the independent auditors’ audit plan - discuss scope, staffing, locations,
reliance upon management and general audit
approach.
|
|
|
(v)
|
Consider
the independent auditors’ judgments about the quality and appropriateness
of the Company’s accounting principles as applied in its financial
reporting.
|
|
|
(vi)
|
Prior
to releasing the year-end results, discuss the results of the audit
with
the external auditors. Discuss certain matters required to be
communicated to audit committees in accordance with the standards
established by the Canadian Institute of Chartered
Accountants.
|
|
|
(vii)
|
Review
and approve the Company’s hiring policies regarding partners, employees
and former partners and employees of the present and former independent
auditors of the Company.
|
|
|
(c)
|
Ethical
and Legal Compliance
|
|
|
(i)
|
On
at least an annual basis, review with the Company’s counsel, any legal
matters that could have a significant impact on the organization’s
financial statements, the Company’s compliance with applicable laws and
regulations, and inquiries received from regulators or governmental
agencies.
|
-
48
-
|
|
(ii)
|
Perform
any other activities consistent with this Charter, the Company’s by-laws,
and governing law, as the Audit Committee or the Board deems necessary
or
appropriate. In particular, the Audit Committee has the
authority to engage independent counsel and other advisers, as
it
determines necessary to carry out its duties. The Company will
provide for appropriate funding, as determined by the Audit Committee,
in
its capacity as a committee of the Board, for payment of (i) compensation
to the independent auditor engaged for the purpose of preparing
or issuing
an audit report or performing other audit, review or attest services
for
the Company, (ii) compensation to any advisers employed by the
Audit
Committee, and (iii) ordinary administrative expenses of the Audit
Committee that are necessary or appropriate in carrying out its
duties.
|
|
|
(d)
|
Whistle
Blowing
|
The
Audit Committee shall put in place procedures for:
|
|
(i)
|
The
receipt, retention, and treatment of complaints received by the
Company
regarding accounting, internal accounting controls, or auditing
matters;
and
|
|
|
(ii)
|
The
confidential, anonymous submission by employees of the Company
of concerns
regarding questionable accounting or auditing
matters.
|
|
|
(e)
|
Other
Audit Committee
Responsibilities
|
|
|
(i)
|
Create
an agenda for the ensuing year.
|
|
|
(ii)
|
Describe
in the Company’s annual information form the Audit Committee’s composition
and responsibilities and how they were discharged in accordance
with the
requirements of MI 52-110F1.
|
|
|
(iii)
|
Submit
the minutes of all meetings of the Audit Committee to the
Board.
|
-
49
-
Appendix
“A”
Non-Audit
Services Request Form
LORUS
THERAPEUTICS INC.
The
Audit Committee approves all audit fees and terms and all non-audit services
provided by the independent auditor and consider whether these services are
compatible with the auditor’s independence. Any member of the Audit
Committee, subject to appropriate delegation, may approve additional proposed
non-audit services that arise between Audit Committee meetings provided that
the
decision to approve the service is presented at the next scheduled Audit
Committee meeting. This form documents the member’s approval of the
non-audit service in a form suitable for distribution at meetings of the
Audit
Committee.
Request
Made By
|
Name,
Title, Date:
|
Detailed
Description of Non-Audit Service Requested(including a general
description of the nature of the services that may make up the
project)
|
|
Engagement
Fee or Range of Fees for this Service
|
|
Prohibited
Services
In
this section please confirm that these services are not “prohibited services”
under section 201 of the Sarbanes-Oxley Act of 2002 and other related rules
or
regulations.
|
These
services would not be considered prohibited
services
|
Issues
considered in forming the conclusion above that should be considered by the
Audit Committee
|
|
-
50
-
Compatibility
with Auditors’ Independence
In
this section please state whether these services are compatible with the
auditors’ independence.
|
These
services are compatible with the auditors’
independence
|
Issues
considered in forming the conclusion above that should be considered by the
audit committee
|
|
Management
Approval
This
form must be reviewed and approved by one authorized member of management
(either the CEO, CFO or Director of Finance before submitting this form to
an
Audit Committee member for final approval.
|
Name,
Title, Date:
|
Audit
Committee Member Approval
|
Name,
Date:
|
-
51
-