SEC 1852(02-2002) Persons who potentially are to respond to the collection of information contained in this form are not required to respond unless the form displays a currently valid OMB control number.
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 20-F
(Mark One)
| o | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934 For the fiscal year ended May 31, 2002 |
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to |
Commission file number 0-19763
LORUS THERAPEUTICS INC.
NOT APPLICABLE
ONTARIO, CANADA
2 MERIDIAN ROAD, TORONTO, ONTARIO M9W 4Z7
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class | Name of each exchange on which registered | |
| NONE | ||
Securities registered or to be registered pursuant to Section 12(g) of the Act.
COMMON SHARES (NO PAR VALUE)
NONE
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
NONE
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
As of December 6, 2002, the Company had 144,422,262 common shares outstanding.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
| Yes x | No o |
Indicate by check mark which financial statement item the registrant has elected to follow.
| Item 17 x | Item 18 o |
2
TABLE OF CONTENTS
PART I |
|||||
Item 1. Identity of Directors, Senior Management and Advisers |
3 | ||||
Item 2. Offer Statistics and Expected Timetable |
3 | ||||
Item 3. Key Information |
3 | ||||
Item 4. Information on the Company |
10 | ||||
Item 5. Operating and Financial Review and Prospects |
18 | ||||
Item 6. Directors, Senior Management and Employees |
37 | ||||
Item 7. Major Shareholders and Related Party Transactions |
51 | ||||
Item 8. Financial Information |
51 | ||||
Item 9. The Offer and Listing |
52 | ||||
Item 10. Additional Information |
54 | ||||
Item 11. Quantitative and Qualitative Disclosures About Market Risk |
61 | ||||
Item 12. Description of Securities Other than Equity Securities |
61 | ||||
PART II |
|||||
Item 13. Defaults, Dividend Arrearages and Delinquencies |
61 | ||||
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds |
61 | ||||
Item 15. [Reserved] |
61 | ||||
Item 16. [Reserved] |
62 | ||||
PART III |
|||||
Item 17. Financial Statements |
62 | ||||
Item 18. Financial Statements |
80 | ||||
EXHIBITS |
|||||
Item 19. Exhibits |
84 | ||||
PART I
Item 1. Identity of Directors, Senior Management and Advisers
Not applicable to Form 20-F filed as an Annual Report.
Item 2. Offer Statistics and Expected Timetable
Not applicable to Form 20-F filed as an Annual Report.
Item 3. Key Information
The financial statements of Lorus Therapeutics Inc. (“Lorus” or the “Company”) have been prepared in accordance with generally accepted accounting principles (“GAAP”) as applied in Canada and in all material respects comply with U.S. GAAP. The Company also identifies significant differences between Canadian and United States GAAP in note 13 to the consolidated financial statements. All amounts are expressed in Canadian dollars unless otherwise noted. Annual references are to Lorus’s fiscal years which end on May 31.
3
Currency Exchange Rates
Lorus’s accounts are maintained in Canadian dollars. In this Annual Report, all dollar amounts are expressed in Canadian dollars except where otherwise indicated.
The following table sets out, for the periods indicated, the high and low noon buying rates in New York City for cable transfers in foreign currencies, the average of such exchange rates on the last day of each month during the periods, and the end period rates, for Canadian dollars expressed in United States dollars, as certified for customs purposes by the Federal Reserve Bank of New York:
| Year End | Year End | Year End | Year End | Year End | ||||||||||||||||
| May 31, 2002 | May 31, 2001 | May 31, 2000 | May 31, 1999 | May 31, 1998 | ||||||||||||||||
Period End |
0.6547 | 0.6468 | 0.6677 | 0.6791 | 0.6863 | |||||||||||||||
Average |
0.6380 | 0.6602 | 0.6794 | 0.6626 | 0.7092 | |||||||||||||||
High |
0.6622 | 0.6831 | 0.6969 | 0.6891 | 0.7305 | |||||||||||||||
Low |
0.6200 | 0.6333 | 0.6607 | 0.6341 | 0.6832 | |||||||||||||||
As at December 6, 2002, the noon buying rate in New York City for Canadian dollars expressed in United States dollars, as certified for customs purposes by the Federal Reserve Bank of New York, was 0.6386.
A. Selected Financial Data
Consolidated Statement of Loss and Deficit Data
For the five most recent financial years ended May 31
| In thousands of Canadian dollars except per share amount | 2002 | 2001 | 2000 | 1999 | 1998 | |||||||||||||||
Net sales or operating revenues |
— | — | — | — | — | |||||||||||||||
Net income (loss) |
(13,487 | ) | (15,213 | ) | (8,599 | ) | (4,623 | ) | (4,742 | ) | ||||||||||
Loss from continuing
operations per share |
(0.09 | ) | (0.11 | ) | (0.10 | ) | (0.12 | ) | (0.13 | ) | ||||||||||
Basic and fully diluted net
loss from operations per share |
(0.09 | ) | (0.11 | ) | (0.10 | ) | (0.12 | ) | (0.13 | ) | ||||||||||
Weighted average number of
shares used in computing basic
and fully diluted net loss per
share (in thousands) |
143,480 | 140,776 | 86,121 | 37,858 | 36,567 | |||||||||||||||
4
Consolidated balance sheet data
For the five most recent financial years ended May 31
| In thousands of Canadian dollars except per share amount | 2002 | 2001 | 2000 | 1999 | 1998 | |||||||||||||||
Total assets |
47,572 | 61,807 | 72,363 | 3,250 | 5,917 | |||||||||||||||
Net assets |
44,140 | 55,942 | 68,755 | 1,920 | 4,785 | |||||||||||||||
Capital stock |
119,009 | 117,324 | 114,924 | 39,490 | 37,732 | |||||||||||||||
B. Capitalization and Indebtedness
Not applicable to Form 20-F filed as an Annual Report.
C. Reason for the Offer and Use of Proceeds
Not applicable to Form 20-F filed as an Annual Report.
D. Risk Factors
Forward Looking Statements
This and other sections of this annual report on Form 20-F (the “Annual Report”) contain forward-looking statements, reflecting the Company’s current expectations regarding future events. Readers are cautioned that these forward-looking statements involve risks and uncertainties, including, without limitation, changing market conditions, the successful and timely completion of clinical studies, the impact of competitive products and pricing, new product development, uncertainties related to the regulatory approval process, product development delays, the Company’s ability to attract and retain business partners, future levels of government funding and the Company’s ability to obtain the capital required for research, product development, operations and marketing. These factors should be carefully considered and readers should not place undue reliance on the Company’s forward-looking statements. Actual events may differ materially from the Company’s current expectations due to risks and uncertainties. Certain of the risks and uncertainties are discussed in the section entitled “Risk Factors”.
Lorus’s statements of “belief” are based upon its results derived to date from its research and development program with animals and its clinical trials in humans and upon which the Company believes there is a reasonable scientific basis to expect the particular results to occur. It is not possible to predict, based upon studies in animals, whether a new therapeutic will be proved to be safe and effective in humans. There can be no assurance that the particular result expected by the Company will occur. Except as required by law, the Company undertakes no obligation to update publicly any forward-looking statements for any reason after the date of this Annual Report or to conform these statements to actual results or to changes in the Company’s expectations.
5
THE COMPANY’S BUSINESS IS SUBJECT TO A NUMBER OF RISK FACTORS THAT ARE SET OUT BELOW:
No Assurance of Successful Development
Lorus has not produced or commercially marketed any product. Although the Company has entered into an agreement in respect of the commercial sale of Virulizin® in Mexico, there can be no assurance that any sales of the product will be made. There can be no assurance that the Company will ever successfully develop commercial products, that the Company will ever achieve significant revenues from such products if they are successfully developed, or that the Company will ever be profitable.
Lack of Operating Profits
Lorus commenced its research and development activities in 1986. To date, the Company has not earned any operating revenue, generated positive cash flow or earned operating profits, and the Company expects to incur significant operating losses for the foreseeable future. To date, there have been no revenues recorded from the sale of any products. Lorus expects to accumulate losses as it continues its product research and development and continues its clinical trials and applies for regulatory approval for the sale of its products. The Company expects to continue to incur substantial operating losses unless and until such time as product sales and royalty payments generate sufficient revenues to fund its continuing operations. The Company expects to have quarter-to-quarter fluctuations in revenues, expenses and losses, some of which could be significant. The Company’s ability to achieve a profitable level of operations is dependent, in large part, on completing product development, obtaining regulatory approvals, and commercializing its products. There can be no assurance that the Company will ever achieve a profitable level of operations.
Liquidity and Capital Requirements
As at May 31, 2002, the Company’s current assets exceeded current liabilities by $35.6 million. Lorus will require substantial additional funds for clinical trials. The Company may seek to obtain additional funds for these purposes through public or private equity or debt financings, collaborative arrangements with pharmaceutical companies and/or from other sources. Any such financing may have a dilutive effect on the holdings of shareholders. There can be no assurance that additional funding will be available at all or on acceptable terms to permit successful commercialization of the Company’s products.
Patents and Proprietary Technology
The Company’s success depends in part on its ability to obtain patents, maintain trade secret protection and operate without infringing the proprietary rights of third parties. The Company has filed patent applications in Canada, the United States and internationally. Other than the patents granted by the patent offices in Canada, the United States and internationally to date, there can be no assurance that these patent applications will be allowed, that the Company will develop additional proprietary products that are patentable, that patents that have already been granted to the Company will provide it with any competitive advantage or will not be challenged by any third parties, or that patents of others will not have an adverse effect on the Company’s ability to do business. Furthermore, there can be no assurance that others will not independently develop similar products, duplicate any of the Company’s products, or design around the products patented, if any, by the Company. In addition, the Company may be required to obtain licences under patents or other proprietary rights of third parties. There can be no assurance that any licences required under such patents or proprietary rights will be available on terms acceptable to the Company. If Lorus does not obtain such licences,
6
it could encounter delays in introducing one or more of its products to the market, while it attempts to design around third party patents, or the Company could find that the development, manufacturing or sale of products requiring such licences could be foreclosed. In addition, Lorus could incur substantial costs in defending suits brought against it on patents or in suits in which it attempts to enforce its own patents, if any, against other parties.
Until such time as further patents are issued to the Company, its ability to maintain the confidentiality of its technology is crucial to its ultimate possible commercial success. While the Company has adopted procedures designed to protect the confidentiality of its technology, there can be no assurance that such arrangements will be effective, that third parties will not gain access to the Company’s trade secrets or disclose the technology, or that the Company can meaningfully protect its rights to its trade secrets. Further, by seeking patent protection in various countries, it is inevitable that a substantial portion of the Company’s technology will become available to its competitors, through publication of such patent applications. In addition, the Company uses contract manufacturers to manufacture its products. In order for these manufacturers to produce the Company’s products, the Company must provide them with valuable confidential information regarding their formulation. If the Company is unable to maintain the confidentiality of its technology, this could have a material adverse impact on the Company’s business and financial condition.
Dependence on the Successful Outcome of Clinical Trials
In order to commercialize its products, Lorus must obtain approval from governmental regulatory agencies. Regulatory approval can take several years and can involve substantial expenditures. There can be no assurance that difficulties or excessive costs will not be encountered by the Company in its efforts to secure necessary approvals or licences, which could delay or prevent the Company from marketing its products. There can be no assurance that the Company will ever obtain necessary regulatory approvals or licences for any of its products.
Before obtaining regulatory approval for the sale of the Company’s products, they must be subjected to extensive preclinical and clinical testing to demonstrate their safety and efficacy for humans. Lorus’s success will depend on the success of its currently ongoing clinical trials and subsequent clinical trials that have not yet begun. There are a number of difficulties and risks associated with clinical trials. The possibility exists that:
| • | the Company may discover that a product candidate does not exhibit the expected therapeutic results in humans; | ||
| • | results may not be statistically significant or predictive of results that will be obtained from large-scale, advanced clinical trials; | ||
| • | patient recruitment may be slower than expected; | ||
| • | the Company or the United States Food and Drug Administration (FDA) or the Health Products and Food Branch of Health Canada (HPFB) may suspend the clinical trials of the Company’s product candidates; | ||
| • | the Company may discover that a product may cause harmful side effects; and | ||
| • | patients may drop out of Lorus’s clinical trials. |
7
Given the uncertainty surrounding the regulatory and clinical trial process, the Company may not be able to develop safe, commercially viable products. If Lorus is unable to successfully develop and commercialize any of its products, this would severely harm its business, financial condition and results of operations and have an adverse impact on the Company’s share price.
Reliance on Clinical Investigators and Contract Research Organizations
Lorus depends on independent clinical investigators and contract research organizations to conduct its clinical trials. Contract research organizations also assist it in the collection and analysis of the Company’s data. These investigators and contract research organizations are not the Company’s employees and it cannot control, other than by contract, the amount of resources, including time, that they devote to the Company’s products. If independent investigators fail to devote sufficient resources to the development of the Company’s products, or if their performance is substandard, it will delay the approval and commercialization of the Company’s products. Further, the FDA and the HPFB require that the Company comply with standards, commonly referred to as “good clinical practice”, for conducting, recording and reporting clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial subjects are protected. If the Company’s independent clinical investigators and contract research organizations fail to comply with good clinical practice, the results of its clinical trials could be called into question and the clinical development of the Company’s products could be delayed. Failure of clinical investigators and contract research organizations to meet their obligations to the Company or comply with good clinical practice procedures could adversely affect the clinical development of Lorus’s products, and have a material adverse effect on its business and financial condition.
Reliance on Contract Manufacturing
The Company currently relies upon contract manufacturers to manufacture small quantities of its products for use in clinical trials. The Company intends to continue to rely upon these and other third parties to manufacture its products in the future. Lorus has limited manufacturing experience and no manufacturing facilities. If Lorus is unable to obtain new or retain its current contract manufacturers, it will not be able to effectively conduct its clinical trials or ultimately commercialize its products. In addition, the Company’s current and any future contract manufacturers may not comply with government regulations, or other regulatory requirements relating to the manufacturing of its products, including compliance with “good manufacturing practice”, or GMP. GMP requires that the methods, facilities or controls used for a drug product’s manufacture, processing, packing or holding follow rules and guidelines to meet certain safety requirements and to ensure the drug has the characteristics and strength the Company claims it has, and meets quality and purity requirements. The risks associated with the Company’s reliance on contract manufacturers include the following:
| • | Contract manufacturers may encounter difficulties in achieving volume production, quality control and quality assurance, and also may experience shortages in qualified personnel. As a result, the Company’s contract manufacturers might not be able to meet its clinical development schedules or adequately manufacture the Company’s products in commercial quantities when required. |
8
| • | Switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult for the Company to find a replacement manufacturer quickly or on terms acceptable to it, or at all. | ||
| • | The Company’s contract manufacturers may not remain in the contract manufacturing business for the time required to successfully produce, store and distribute its products. | ||
| • | The Company’s contract manufacturers are subject to ongoing periodic, unannounced inspection by the FDA and the HPFB and corresponding governmental agencies to ensure strict compliance with, among other things, GMP, in addition to other governmental regulations and corresponding foreign standards. Lorus does not have control over, other than by contract, third party manufacturers’ compliance with these regulations and standards. | ||
| • | If Lorus needs to change manufacturers, the FDA and the HPFB and corresponding regulatory agencies must approve these manufacturers in advance, which will involve testing and additional inspections to ensure compliance with these regulations and standards. |
The Company’s dependence on third parties for the manufacture of its products may have an adverse impact on its ability to develop and deliver products on a timely and competitive basis and may have a material adverse effect on its business and financial condition.
Sales, Marketing and Distribution Capabilities
Lorus does not have any sales, marketing or distribution capabilities. In order to commercialize its products, if any are approved, the Company must either acquire or internally develop sales, marketing and distribution capabilities or make arrangements with third parties to perform these services for it. If Lorus obtains regulatory approval for its existing products, it intends to rely on relationships with one or more pharmaceutical companies or other third parties with established distribution systems and direct sales forces to market its products. If the Company decides to market any of its products directly, it must either acquire or internally develop a marketing and sales force with technical expertise and with supporting distribution capabilities. The acquisition or development of a sales and distribution infrastructure would require substantial resources, which may divert the attention of management and key personnel, and have a negative impact on the Company’s product development efforts. Moreover, Lorus may not be able to establish in-house sales and distribution capabilities or relationships with third parties. The inability to market its products could have a material adverse effect on the Company’s business and financial condition.
Technology and Competition
Lorus’s success depends on developing and maintaining a competitive position in the development of products and technologies in its area of expertise. Biotechnology is a rapidly evolving field in which new developments are expected to continue at a fast pace. Technological competition in the industry from other biotechnology companies and others diversifying into the field is intense and expected to increase. Many of these companies have substantially greater research and development capability and experience, and marketing, financial and managerial resources than Lorus does, and represent significant long-term competition for Lorus. The acquisition of competing biotechnology companies by large There can be no assurance that
9
developments by others will not render Lorus’s products or technologies non-competitive or obsolete, or that Lorus will be able to achieve the level of acceptance within the medical community necessary to compete successfully. Lorus is aware of several potential competitors that are at various stages of development or that have commercial sale of products that may address similar cancer indications. The success of the Company’s competitors and their products may have a material adverse impact on Lorus’s business and financial condition.
Product Liability and Insurance
The clinical testing and proposed commercialization of Virulizin® and the clinical testing of GTI-2040 and GTI-2501 involve risks of product liability as well as patient injury. While the Company currently maintains limited product liability insurance, there can be no assurance that product liability insurance will continue to be available to the Company on commercially reasonable terms. Product liability claims might also exceed the amounts of such coverage. If Lorus is unable to obtain insurance, is unable to obtain insurance on commercially reasonable terms or if any claims against Lorus exceed the amounts of its insurance coverage, this could have a material adverse impact on Lorus’s business and financial condition.
Dependence on Key Personnel
The Company’s success depends in large part upon its ability to attract and retain highly qualified scientific and management personnel. Lorus faces competition for such personnel from other companies, academic institutions, government entities and other organizations. There can be no assurance that the Company will retain its current personnel and will be able to continue to attract qualified personnel.
Volatility of Share Price
Historically, the market price for securities of biopharmaceutical companies, including Lorus’s, has been volatile. Future announcements concerning Lorus or its competitors, including the results of testing, technological innovations or commercial products, government regulations, developments concerning proprietary rights, litigation and public concerns as to the safety of the Company’s products may have a significant impact on the market price of its common shares. In addition, the Company’s share price may be affected by sales by existing shareholders.
Dividend Policy
The Company has never paid dividends on its common shares and does not anticipate paying dividends on its common shares in the foreseeable future.
Item 4. Information on the Company
A. History and Development of the Company
Lorus Therapeutics Inc. was incorporated under the Business Corporations Act (Ontario) on September 5, 1986 under the name RML Medical Laboratories Inc. On October 28, 1991, RML Medical Laboratories Inc. amalgamated with Mint Gold Resources Ltd., resulting in the Company becoming a reporting issuer (as defined under applicable securities law) in Ontario, on such date. On August 25, 1992, the Company changed its name to IMUTEC Corporation. On November 27, 1996, the Company changed its name to Imutec Pharma Inc., and on November 19, 1998, the Company changed its name to Lorus Therapeutics Inc.
10
The address of the Company’s head and principal office is 2 Meridian Road, Toronto, Ontario, Canada, M9W 4Z7.
Unless otherwise noted or the context otherwise indicates, references to the Company include Lorus together with its subsidiaries. Lorus’s subsidiaries are GeneSense Technologies Inc. (“GeneSense”), a company incorporated under the laws of Canada of which the Company owns 100% of the issued and outstanding share capital and NuChem Pharmaceuticals Inc. (“NuChem”), a company incorporated under the laws of Ontario of which Lorus owns 80% of the issued and outstanding share capital.
Certain terms not otherwise defined in this Annual Report are found in the Glossary, located at pages 80 through 82 of this Annual Report.
B. Business Overview
Lorus is a biopharmaceutical company focused on the research and development of cancer therapies. Lorus’s goal is to capitalize on its research, pre-clinical, clinical and regulatory expertise by developing new drug candidates that can be used, either alone or in combination, to successfully manage cancer. Through its own discovery efforts and an in-licensing and acquisition program, the Company is building a portfolio of promising anti-cancer drugs. With products entering all stages of evaluation, from pre-clinical through Phase III trials, the Company is a leader in the development of therapeutics that complement the new cancer treatment paradigm that seeks to manage the disease with efficacious, non-toxic compounds that improve patients’ quality of life.
Lorus seeks to reduce the risk associated with individual products or single technology platforms by pursuing a wide variety of promising anti-cancer compounds derived from different platform technologies. Lorus intends to develop compounds that are efficacious and have low-toxicity, ensuring the drugs will be well tolerated by patients and will be able to be tested in combination with other approved compounds on the market.
From January 1987 to December 1997, the Company focused its efforts solely on the development of VIRULIZIN®, a potential new drug functioning as a biologic response modifier for the treatment of cancer. In November 1997, the Company sub-licensed, on an exclusive world-wide basis until the later of patent expiry or marketing approval, from Ion Pharmaceuticals, Inc. (“Ion”) (a wholly-owned subsidiary of Sheffield Pharmaceuticals, Inc.) analogues of clotrimazole (“CLT”), a molecule with anti-angiogenic and anti-proliferative properties, for anti-cancer indications as well as actinic keratosis.
On October 29, 1999, Lorus acquired all of the issued and outstanding shares of GeneSense (the “GeneSense Acquisition”), a private biopharmaceutical company that specializes in developing novel oligonucleotide therapeutics for cancer and infectious diseases. Pursuant to the GeneSense Acquisition, the Company obtained two anti-cancer products in late-stage, pre-clinical development, in addition to several other products in the research stage. Lorus believes that the GeneSense Acquisition also added depth to its research and development capabilities.
As a consequence of the GeneSense Acquisition, Lorus now holds an exclusive worldwide license from the University of Manitoba and Cancer Care Manitoba (formerly The Manitoba Cancer Treatment and Research Foundation) to develop certain oligonucleotide technologies. Antisense technology, one of the oligonucleotide technologies, works at the genetic level to interrupt the process by which disease-causing proteins are produced in order to treat a wide range of diseases, including cancer and infectious diseases.
11
Business Strategy
By developing cancer therapeutics using different mechanisms of action that may be efficacious against a wide variety of cancers, the Company seeks to maximize its opportunity to address multiple cancer therapeutic markets. In its efforts to obtain the greatest return on its investment in each drug candidate, the Company separately evaluates the merits of each candidate throughout the clinical trial process and considers commercialization opportunities when appropriate. The Company intends to partner with pharmaceutical companies for the sales, marketing and distribution of products. See “Co-development, Marketing and Distribution.”
Production and Testing
Preclinical testing and certain research and development work has been performed at various contract laboratories. Clinical trials have been undertaken at various medical centers in Canada, the United States and Mexico.
Manufacturing
On May 12, 1998, the Company signed an agreement with Torcan Chemicals for the manufacture of five NuChem Analogues. The compounds were synthesized in sufficient quantity for use in the pre-clinical toxicity and efficacy studies.
The Company has entered into a contract with Proligo LLC, a cGMP manufacturer, to produce its bulk active drug substance for its antisense compounds. The manufacturer supplied bulk active drug for Good Laboratory Practices (GLP) toxicology studies and drug stability studies and has supplied bulk active drug, subsequently formulated, for both the GTI-2040 and GTI-2501 clinical trials. Proligo has filed a drug master file (DMF) with the FDA and have supplied the necessary documentation to support the IND submission.
In March 2001, the Company signed an agreement with Dalton Chemical Laboratories Inc. for the manufacturing of its immunotherapeutic compound VIRULIZIN®. The drug is being manufactured for the Phase III clinical trial program that the Company initiated in fiscal 2002 and for the planned supply of VIRULIZIN® for the Company’s licensee, Mayne Pharma Inc. (formerly Faulding Canada Inc.) for malignant melanoma treatment in Mexico.
Intellectual property and Protection of Confidential Information and Technology
The Company regards its issued patents and pending applications as important in establishing and maintaining a competitive position with respect to its products and technology. As at November 30, 2002, the Company owns or has rights under 40 issued or pending patents in Canada and the United States as well as a number of other pending patent applications in multiple jurisdictions.
With regard to antisense compounds, the Company has protected its intellectual property rights by, among other things, filing patent applications with respect to intellectual property considered important to the development of its business. The Company also relies upon trade secrets, unpatented know-how and continuing technology innovation to develop and maintain its competitive position.
12
There can be no assurance, however, that pending applications will result in issued patents, or that issued patents will be held valid and enforceable if challenged, or that a competitor will not be able to circumvent any such issued patents by adoption of a competitive, though non-infringing product or process. Interpretation and evaluation of pharmaceutical or biotechnology patent claims present complex and often novel legal and factual questions. The Company’s business could be adversely affected by increased competition in the event that any patent granted to it is held to be invalid or unenforceable or is inadequate in scope to protect the Company’s operations.
While the Company believes that its products and technology do not infringe proprietary rights of others, there can be no assurance that third parties will not assert infringement claims in the future or that such claims will not be successful. Furthermore, the Company could incur substantial costs in defending itself against patent infringement claims brought by others or in prosecuting suits against others.
In addition, no assurance can be given that others will not obtain patents that the Company would need to license, or that if a licence is required that it would be available on reasonable terms, or that if a licence is not obtained that the Company would be able to circumvent, through a reasonable investment of time and expense, such outside patents. Whether the Company obtains a licence would depend on the terms offered, the degree of risk of infringement, the vulnerability of the patent to invalidation, and the ease of circumventing the patent.
Until such time, if ever, that further patents are issued to the Company, the Company will rely upon the law of trade secrets to the extent possible given the publication requirements under international patent treaty laws and/or requirements under foreign patent laws to protect its technology and the products incorporating the technology. In this regard, the Company has adopted certain confidentiality procedures. These include: limiting access to its confidential information to certain key personnel; requiring all of its directors, officers, employees and consultants and others who may have access to the Company’s intellectual property to enter into confidentiality agreements which prohibit the use of or disclosure of confidential information to third parties; and implementing physical security measures designed to restrict access to such confidential information and products. The ability of the Company to maintain the confidentiality of its technology is crucial to its ultimate possible commercial success. No assurance can be given that the procedures adopted by the Company to protect the confidentiality of its technology will be effective, that third parties will not gain access to Registrant’s trade secrets or disclose the technology, or that the Company can meaningfully protect its rights to its technology. Further, by seeking the aforementioned patent protection in various countries, it is inevitable that a substantial portion of the Company’s technology will become available to its competitors, through publication of such patent applications.
License Agreement
The University of Manitoba (the “University”), Cancer Care Manitoba (formerly the Manitoba Cancer Treatment and Research Foundation) (“Cancer Care”), Dr. Jim Wright and Dr. Aiping Young entered into an exclusive license agreement (the “License Agreement”) with GeneSense dated June 20, 1997 pursuant to which GeneSense was granted an exclusive world-wide license to certain patent rights with the right to sub-license. In consideration for the exclusive license to GeneSense of the patent rights, the University and Cancer Care are entitled to an aggregate of 1.67% of the net sales received by GeneSense from the sale of products or processes derived from the patent rights and 1.67% of all monies received by GeneSense from sub-licenses of the patent rights. GeneSense is solely responsible for the preparation, filing, prosecution and maintenance of all patent applications and patents included in the patent rights and all related expenses. Pursuant to the terms of
13
the License Agreement, any and all improvements to any of the patent rights derived in whole or in part by GeneSense after the date of the License Agreement are not included within the scope of the License Agreement and do not trigger any payment of royalties.
Regulatory Requirements
Regulation by government authorities in Canada, the United States, Mexico and the European Union is a significant factor in the current research and drug development activities of the Company. In order to clinically test, manufacture and market drug products for therapeutic use, the Company must satisfy the rigorous mandatory procedures and standards established by the regulatory agencies in the countries in which it currently operates or intends to operate.
The laws of most of these countries require the licensing of manufacturing facilities, carefully controlled research and the extensive testing of products. Biotechnology companies must establish the safety and efficacy of their new products in clinical trials, as well as satisfy all regulatory requirements. The safety and efficacy of a new drug must be shown through clinical trials of the drug carried out in accordance with the mandatory procedures and standards established by regulatory agencies.
Regulatory compliance can take several years and can involve substantial expenditures. There can be no assurance that difficulties or excessive costs will not be encountered by the Company in its efforts to secure necessary approvals, which could delay or prevent the Company from manufacturing or marketing its products.
Canada
In Canada, the manufacture and sale of new drugs are controlled by the Therapeutic Products Programme (“TPP”). New drugs must pass through a number of testing stages, including pre-clinical testing and clinical trials. Preclinical testing involves testing the new drug’s chemistry, pharmacology and toxicology in vitro and in animals. Successful results (that is, potentially valuable pharmacological activity combined with an acceptable low level of toxicity) enable the manufacturer of the new drug to file an IND submission to begin clinical trials involving humans.
In order to study a drug in Canadian patients, an IND submission must be filed with the TPP. The IND submission must contain specified information, including the results of the pre-clinical tests completed at the time of the submission and any available information regarding use of the drug in humans. In addition, since the method of manufacture may affect the efficacy and safety of a new drug, information on manufacturing methods and standards and the stability of the substance and dosage form must be presented. Production methods and quality control procedures must be in place to ensure an acceptably pure product, essentially free of contamination, and to ensure uniformity with respect to all quality aspects.
Provided the TPP does not reject an IND submission, clinical trials can begin. Clinical trials are carried out in three phases or a combination thereof. Phase I involves studies to evaluate toxicity in humans. The new drug is administered to human patients who have met the clinical trial entry criteria to determine pharmacokinetics, human tolerance and prevalence of adverse side effects. Phases II and III involve therapeutic studies. In Phase II, efficacy, dosage, side effects and safety are established in a small number of patients who have the disease or disorder that the new drug is intended to treat. In Phase III, there are controlled clinical trials in which the new drug is administered to a large number of patients who are likely to receive benefit from
14
the new drug. In Phase III, the effectiveness of the new drug is compared to that of standard accepted methods of treatment in order to provide sufficient data for the statistical proof of safety and efficacy for the new drug.
If clinical studies establish that a new drug has value, the manufacturer submits a NDS application to the TPP for marketing approval. The NDS contains all information known about the new drug, including the results of pre-clinical testing and clinical trials. Information about a substance contained in an NDS includes its proper name, its chemical name, details on its method of manufacturing and purification and its biological, pharmacological and toxicological properties. The NDS also provides information about the dosage form of the new drug, including a quantitative listing of all ingredients used in its formulation, its method of manufacture, packaging and labelling, the results of stability tests, and its diagnostic or therapeutic claims and side effects, as well as details of the clinical trials to support the safety and efficacy of the new drug. All aspects of the NDS are critically reviewed by the TPP. If an NDS is found satisfactory, a notice of compliance is issued permitting the new drug to be sold.
The TPP has a policy of priority evaluation of new drug submissions for all drugs intended for serious or life-threatening diseases for which no drug product has received regulatory approval in Canada and for which there is reasonable scientific evidence to indicate that the proposed new drug is safe and may provide effective treatment.
The monitoring of a new drug does not cease once it is on the market. For example, a manufacturer of a new drug must report any new information received concerning serious side effects, as well as the failure of the new drug to produce desired effects. As well, if the TPP determines it to be in the interest of public health, a notice of compliance for a new drug may be suspended and the new drug may be removed from the market.
An exception to the foregoing requirements relating to the manufacture and sale of a new drug is the limited authorization that may be available in respect of the sale of new drugs for emergency treatment. Under the special access program, the TPP may authorize the sale of a quantity of a new drug for human use to a specific practitioner for the emergency treatment of a patient under the practitioner’s care. Prior to authorization, the practitioner must supply the TPP with information concerning the medical emergency for which the new drug is required, such data as is in the possession of the practitioner with respect to the use, safety and efficacy of the new drug, the names of the institutions at which the new drug is to be used and such other information as may be requested by the TPP. In addition, the practitioner must agree to report to both the drug manufacturer and the TPP the results of the new drug’s use in the medical emergency, including information concerning adverse reactions, and must account to the TPP for all quantities of the new drug made available.
The Canadian regulatory approval requirements for new drugs outlined above are similar to those of other major pharmaceutical markets. While the testing carried out in Canada is often acceptable for the purposes of regulatory submissions in other countries, supplementary testing may be requested by individual regulatory authorities during their assessment of any submission. There can be no assurance that the clinical testing conducted under the TPP authorization or the approval of regulatory authorities of other countries will be accepted by regulatory authorities outside Canada or such other countries.
15
United States
In the United States, the manufacture and sale of new drugs are controlled by the FDA. New drugs require FDA approval of a marketing application (e.g., an NDA or product licence application) prior to commercial sale. To obtain marketing approval, data from adequate and well-controlled clinical investigations, demonstrating to the FDA’s satisfaction a new drug’s safety and effectiveness for its intended use, are required. Such data are generated in studies conducted pursuant to an IND submission, similar to that required in Canada. As in Canada, clinical studies are characterized as Phase I, Phase II and Phase III trials or a combination thereof. In a marketing application, the manufacturer must also demonstrate the identity, potency, quality and purity of the active ingredients of the new drug involved, and the stability of those ingredients. Further, the manufacturing facilities, equipment, processes and quality controls for the new drug must comply with the FDA’s cGMP regulations for drugs or biologic products both in a pre-licensing inspection before product licensing and in subsequent periodic inspections after licensing. In the case of a biologic product, an establishment licence must be obtained prior to marketing and batch releasing.
A five-year period of market exclusivity for a drug comprising a New Chemical Entity (“NCE”) is available to an applicant that succeeds in obtaining FDA approval of a NCE, provided the active ingredient of the NCE has never before been approved in a NDA. During this exclusivity period, the FDA may not approve any abbreviated application filed by another sponsor for a generic version of the NCE. Further, a three-year period of market exclusivity for a new use or indication for a previously approved drug is available to an applicant that submits new clinical studies that are essential to support the new use or indication. During the latter period of exclusivity, the FDA may not approve an abbreviated application filed by another sponsor for a generic version of the product for that use or indication.
The FDA has “fast track” regulations intended to accelerate the approval process for the development, evaluation and marketing of new drugs used to diagnose or treat life-threatening and severely debilitating illnesses for which no satisfactory alternative therapies exist. “Fast track” designation affords early interaction with the FDA in terms of protocol design, and permits, although it does not require, the FDA to issue marketing approval after completion of early stage clinical trials (although the FDA may require subsequent clinical trials or even post-approval efficacy studies).
Mexico
In Mexico, the manufacture and sale of new drugs are controlled by the SSA. The regulatory requirements in Mexico operate under similar regulatory principles as other international jurisdictions.
Summary
The process of completing clinical trials and obtaining regulatory approval for a new drug takes a number of years and require the expenditure of substantial resources. Once a new drug or product licence application is submitted, there can be no assurance that a regulatory agency will review and approve the application in a timely manner. Even after initial approval has been obtained, further studies, including post-marketing studies, may be required to provide additional data on safety necessary to gain approval for the use of the new drug as a treatment for clinical indications other than those for which the new drug was initially tested. Also, regulatory agencies may require post-marketing surveillance programs to monitor a new drug’s side effects. Results of post-marketing programs may limit or expand the further marketing of new drugs. A serious
16
safety or effectiveness problem involving an approved new drug may result in a regulatory agency requiring withdrawal of the new drug from the market and possible civil action.
In addition to the regulatory product approval framework, biotechnology companies, including the Company, are subject to regulation under local provincial, state and federal law, including requirements regarding occupational safety, laboratory practices, environmental protection and hazardous substance control, and may be subject to other present and future local, provincial, state, federal and foreign regulation, including possible future regulation of the biotechnology industry.
Regulatory Strategy
The Company’s overall regulatory strategy is to work with the TPP in Canada, the FDA in the United States, the EMEA in Europe, the SSA in Mexico and any other local regulatory agencies to have drug applications approved for use of VIRULIZIN®, GTI-2040, GTI-2501, and NuChem analogues in clinical trials (alone and/or in combination with chemotherapeutic compounds) and subsequently for sale in international markets. Where possible, the Company intends to take advantage of opportunities for accelerated consideration of drugs designed to treat rare and serious or life-threatening diseases. The Company also intends to pursue priority evaluation of any application for marketing approval filed in Canada, the United States, the European Economic Community or Mexico. The Company also intends to file additional drug applications in other markets where commercial opportunities exist. There can be no assurance that the Company will be able to pursue these opportunities successfully.
Co-Development, Marketing and Distribution
The Company’s objective is to maximize the therapeutic value and potential commercial success of VIRULIZIN®, its antisense technology, and the NuChem analogues. In its efforts to obtain the greatest return on its investment in each drug candidate, the Company separately evaluates the merits of each candidate throughout the development process and will consider commercialization opportunities when appropriate. The Company intends to partner with pharmaceutical companies for the sales, marketing and distribution of the Company’s products.
The Company has a variety of academic partnerships including: Hospital for Sick Children; McGill University; Ontario Cancer Institute; United States NCI; University of Western Ontario, and the University of Chicago Cancer Center.
In July 2000, the Company entered into a five year agreement with AVI BioPharma Inc. (“AVI”) Portland, Oregon, U.S.A. (a leading U.S. company in the area of antisense technology) for the evaluation and co-development of antisense drug therapies for cancer and infectious diseases. Under the terms of the agreement, the Company and AVI will each retain an ownership interest in any jointly developed compound. Drugs discovered together may also be developed independently with royalty payments to the other party.
In September 2001, the Company executed a licence and distribution agreement with Mayne Pharma Inc. (formerly Faulding Canada Inc.). Mayne Pharma will distribute and sell VIRULIZIN® in Mexico for the treatment of malignant melanoma. Under the terms of the agreement, Mayne Pharma has exercised its option to obtain the rights to distribute and sell VIRULIZIN® in Brazil and also for Argentina. The Company will arrange for the manufacture of VIRULIZIN® and receive a royalty on sales.
17
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly evolving technology and intense competition. There are many companies in both these industries that are focusing their efforts on activities similar to those of the Company. Some of these are companies with established positions in the pharmaceutical industry and may have substantially more financial and technical resources, more extensive research and development capabilities, and greater marketing, distribution, production and human resources than the Company. In addition, the Company may face competition from other companies for opportunities to enter into collaborative agreements with biotechnology and pharmaceutical companies and academic institutions. Many of these other companies are not solely focused on cancer, as is the mission of the Company’s drug development. The Company specializes in the development of drugs that will help manage cancer. With products in late stage pre-clinical through to Phase III development, spanning three different platform technologies focused on cancer, the Company believes it has multiple opportunities for success.
Products that may compete with the Company’s include chemotherapeutic agents, monoclonal antibodies, antisense therapies and immunotherapies with novel mechanisms of action. These are drugs that are delivered by specific means and are targeting cancers with large disease populations. The Company also expects it may experience competition from established and emerging pharmaceutical and biotechnology companies that have other forms of treatment for the cancers targeted by the Company. There are many drugs currently in development for the treatment of cancer that employ a number of novel approaches for attacking these cancers. Cancer is a complex disease with more than 100 indications requiring drugs for treatment. The drugs in competition with the Company’s drugs have specific targets for attacking the disease, targets which are not necessarily the same as the Company’s. These competitive drugs therefore could potentially also be used together in combination therapies with the Company’s drugs to manage the disease.
C. Organizational Structure
Lorus’ subsidiaries are GeneSense, a company incorporated under the laws of Canada of which the Company owns 100% of the issued and outstanding share capital and NuChem, a company incorporated under the laws of Ontario of which Lorus owns 80% of the issued and outstanding common share capital.
D. Property, Plants and Equipment
The Company’s head office, which occupies 20,500 square feet, is located at 2 Meridian Road, Toronto, Ontario. The premises include approximately 8,000 square feet of laboratory and research space.
Item 5. Operating and Financial Review and Prospects
A. Operating Results
The following discussion should be read in conjunction with the audited consolidated financial statements and notes prepared in accordance with Canadian generally accepted accounting principles (GAAP). The Company also identifies significant differences between Canadian and United States GAAP in note 13 to the consolidated financial statements. All amounts are expressed in Canadian dollars unless otherwise noted. Annual references are to the Company’s fiscal years which end on May 31.
18
The Company is a biopharmaceutical company specializing in the research, development and commercialization of pharmaceutical products and technologies for the management of cancer. With products in all stages of evaluation, from preclinical through Phase III trials, and a product approved in Mexico for malignant melanoma, the Company is a leader in the development of therapeutics that seek to manage cancer with efficacious non-toxic compounds that improve patients’ quality of life.
The success of the Company depends on the efficacy and safety of its products in clinical trials and on obtaining the necessary regulatory approvals to market its products. The Company believes that the treatment and management of cancer will continue to be addressed through combinations of different therapies. Many cancer drugs currently approved for use are very toxic with severe side effects. The Company is a leader in the development of cancer drugs with low toxicity. Effective drugs with lower toxicity and fewer side effects could have broad application in cancer treatment while improving the quality of life of a patient with cancer.
The Company’s strategy is to pursue the development of drug candidates using several therapeutic approaches, dependent upon different technologies, which mitigates the development risks associated with a single technology platform. The Company’s most advanced anticancer drugs in its pipeline flow from three platform technologies: Immunotherapeutics (Virulizin®); Antisense (GTI compounds); and small molecule or Chemotherapeutics (NuChem compounds).
Critical Accounting Policies
We periodically review our financial reporting and disclosure practices and accounting policies to ensure that our financial reporting and disclosure system provided accurate and transparent information relative to current economic and business environment. As part of this process, we have reviewed our selection, application and communication of critical accounting policies and financial disclosures. We have determined that our critical accounting policies relating to our core ongoing business activities are primarily those that relate to our research and development programs. Other important accounting policies are described in Note 2 to our consolidated financial statements for the year ended May 31, 2002.
Research and Development
The Company incurs research and development costs in connection with advancing its compounds through pre clinical and clinical trials. Research and development costs include internal resource costs as well as external suppliers for the manufacturing of drugs and for the performance of clinical trials and other related activities. Research costs are charged to expense as incurred as required under generally accepted accounting principles. Development costs, including the cost of drugs for use in clinical trials, are expensed as incurred unless they meet the criteria under generally accepted accounting principles for deferral and amortization. To date, no development costs have been capitalized, as the criteria for capitalization have not been met. The Company does not believe that any development costs will be capitalized over the next twelve months given the current stage of our drug development programs and the planned activities for fiscal 2003.
The Company capitalizes costs of acquired research and development from third parties. The Company assesses the nature of the underlying assets acquired to determine if the asset has an indefinite life. If the asset has an indefinite life, the acquired asset is not amortized but is instead tested annually for impairment. If the asset is determined to have a finite life, the acquired asset is amortized over its expected useful life.
19
Results of Operations
The Company has incurred annual operating losses since inception related to the research, manufacturing, and clinical development of its proprietary compounds. The Company has not received any revenue from the sales of products to date. Three products are in the clinical trial stage of development and several potential compounds exist in preclinical studies. Losses will continue as the Company invests in these preclinical research and clinical drug development programs.
Research and Development
Research and development expenditures totaled $8.7 million in 2002 compared to $9.8 million in 2001 and $4.2 million in 2000. The decrease in 2002 from 2001 resulted from the cost of antisense drugs purchased in 2001 which are being used in fiscal 2002 and future years, but were expensed when purchased. This decrease in cost more than offset the increased expenditures in 2002 for an expanded clinical program including the Phase III Virulizin® clinical trial, Phase II GTI-2040 combination chemotherapy trial and Phase I GTI-2501 trial. Regulatory costs were also higher in 2002 due mainly to the initiation of the Phase III clinical trial in the United States. The increase in 2001 over 2000 was due mainly to the cost of antisense and Virulizin® drug development programs, the operating costs of the Company’s research facilities and the amortization of acquired research and development for a full year in 2001 compared to seven months post-acquisition of GeneSense in 2000.
General and Administrative
General and administrative expenses totaled $4.9 million in 2002 compared to $6.4 million in 2001 and $3.7 million in 2000. The decrease in 2002 expenses over 2001 was due mainly to lower spending on patent fees and advisory services as well as lower recruiting costs. The 2001 results include a full year of administration costs related to GeneSense compared to seven months in 2000, with higher costs relating to intellectual property, recruiting and advisory services, and licensing activities.
Depreciation and Amortization
Depreciation and amortization expenses totaled $2.0 million in 2002 compared to $1.9 million in 2001 and $1.2 million in 2000. The increase in 2002 and 2001 over 2000 related primarily to the amortization of goodwill established on the acquisition of GeneSense for twelve months in 2001 and seven months in 2000. Consistent with the application of new accounting pronouncements the Company will not amortize goodwill in future periods and will be assessing acquired intangible assets to determine if continued amortization is appropriate.
Interest Income
Interest income totaled $2.0 million in 2002 compared to $2.9 million in 2001 and $0.5 million in 2000. The decrease in 2002 compared to 2001 was due to lower cash and short-term investment balances in 2002 and the decline in market interest rates. The increase in 2001 over 2000 was due primarily to a higher average cash and investment balance than the previous year. Net cash proceeds of $61.1 million were raised from the issue of common shares and the exercise of warrants in 2000, with $42.0 million of this raised in the last month of 2000.
20
Loss for the Period
The loss for the year totaled $13.5 million in 2002 compared to $15.2 million in 2001 and $8.6 million in 2000. The decrease in 2002 from 2001 was primarily due to reduced spending on general and administrative expenses and net spending reductions on research and development activities due to lower drug purchases partially offset by lower interest income. The increase in 2001 over 2000 resulted mainly from higher clinical development costs, which included higher trial initiation and monitoring costs, manufacturing and regulatory costs in preparation for the Virulizin® phase III trial and antisense drug costs. Additionally, 2001 results included twelve months of research and development costs, amortization of acquired research and development and goodwill, and administration costs related to the GeneSense Acquisition compared to seven months in 2000.
The loss per common share was $0.09 in 2002 compared to $0.11 in 2001 and $0.10 in 2000. The loss per share in each year was comparable although the average number of shares increased significantly in 2001 over 2000.
Financial Summary
The following table summarizes selected unaudited quarterly financial data over the past two fiscal years ended May 31 in each year. The information should be read in conjunction with the Company’s consolidated financial statements and related notes. The operating results for any quarter are not necessarily indicative of results for any future period.
Fiscal 2002
| Fourth | Third | Second | First | |||||||||||||
| Quarter | Quarter | Quarter | Quarter | |||||||||||||
Net loss |
$ | 3,720 | $ | 3,028 | $ | 3,683 | $ | 3,056 | ||||||||
Basic and diluted loss per share |
$ | 0.02 | $ | 0.02 | $ | 0.03 | $ | 0.02 | ||||||||
Fiscal 2001
| Fourth | Third | Second | First | |||||||||||||
| Quarter | Quarter | Quarter | Quarter | |||||||||||||
Net loss |
$ | 6,133 | $ | 2,738 | $ | 3,806 | $ | 2,536 | ||||||||
Basic and diluted loss per share |
$ | 0.04 | $ | 0.02 | $ | 0.03 | $ | 0.02 | ||||||||
B. Liquidity and Capital Resources
Since inception, the Company has financed its operations and technology acquisitions primarily from equity financing, the exercise of warrants and stock options, and interest income on funds held for future investment. The Company believes that its available cash, cash equivalents and short-term investments, and the interest earned thereon, should be sufficient to finance its operations and capital needs for at least the next twelve months.
21
Financing
In 2002, the Company issued common shares on the exercise of stock options and warrants for proceeds of $1.4 million. In 2001, the Company issued common shares on the exercise of warrants and stock options, and under the alternate compensation plan in the aggregate amount of $2.0 million.
In 2000, the Company raised gross proceeds of $64.5 million from two public offerings and the exercise of outstanding warrants, and completed a major acquisition through the issuance of common shares. In October 1999, the Company issued 36,050,000 common shares and converted existing GeneSense warrants to new warrants for the acquisition of GeneSense valued at $14.8 million. These new warrants were exercised in early 2000 for gross proceeds of $5.0 million. Cash paid on the acquisition of GeneSense net of cash received totaled $0.5 million.
In May 2000, the Company issued 15,333,334 common shares at $3.00 per share for gross proceeds of $46.0 million. Additional warrant exercises during 2000 provided an additional $3.5 million in cash proceeds.
Operating Cash Requirements
The Company’s cash burn (cash used in operating activities) totaled $11.9 million in 2002 compared to $9.7 million in 2001 and $5.4 million in 2000. The cash burn increased in 2002 over 2001 mainly due to changes in the timing of accounts payable partially offset by reduced expenditures in operating activities. The cash burn increased in 2001 over 2000 due mainly to a higher level of research and development activity and higher clinical development costs. In 2001 research and development expenses and general and administrative costs increased also due to a full year of costs related to GeneSense activities compared to seven months in 2000.
The Company’s cash burn is expected to increase in 2003 due to increased clinical development activity.
Cash Position
At May 31, 2002 the Company had cash and cash equivalents and short-term investments totaling $37.8 million compared to $48.8 million at the end of 2001. The Company invests in highly rated and liquid government and corporate debt instruments. Investment decisions are made in accordance with an established investment policy administered by senior management and overseen by the Board of Directors.
Working capital (representing primarily cash and cash equivalents and short-term investments) at May 31, 2002 was $35.6 million ($44.5 million in 2001). The Company does not expect to generate a positive cash flow from operations for several years due to substantial additional research and development costs, including costs related to drug discovery, preclinical testing, clinical trials, manufacturing costs and operating expenses associated with supporting these activities. The Company may need to raise additional capital to fund operations over the long-term.
22
The Company intends to raise additional funds through equity financings, collaborative arrangements, acquisitions or otherwise. The Company may seek to access the public or private equity markets from time to time, even if it does not have an immediate need for additional capital at that time.
The Company intends to use its resources to fund its existing drug development programs and develop new programs from its portfolio of preclinical research technologies. The amounts actually expended for research and drug development activities and the timing of such expenditures will depend on many factors, including the progress of the Company’s research and drug development programs, the results of preclinical and clinical trials, the timing of regulatory submissions and approvals, the ability of the Company to establish collaborative research or drug development arrangements with other organizations, the impact of any in-licensed or acquired technologies, the impact from technological advances, determinations as to the commercial potential of the Company’s compounds, and the timing and status of competitive products.
Risks and Uncertainties
Funding needs may vary depending on many factors including: the progress and number of research and drug development programs; costs associated with clinical trials and the regulatory process; costs related to maintaining drug manufacturing sources; costs of prosecuting or enforcing patent claims and other intellectual property rights; collaborative and license agreements with third parties; and opportunities to in-license or acquire new products.
The Company’s interest income is subject to fluctuations due to changes in interest rates in its investment portfolio of debt securities. Investments are held to maturity and have staggered maturities to minimize interest rate risk.
The Company purchases some services and manufactured drugs in U.S. currency, and conducts clinical trials in the United States. U.S. dollar expenditures are expected to increase in 2003 from additional clinical activity in the United States. The Company does not currently engage in hedging its U.S. currency requirements to reduce exchange rate risk, but may do so in the future if conditions warrant.
Recent Accounting Pronouncements
In August 2001, the Canadian Accounting Standards board (“AcSB”) issued Handbook Section 1581, “Business Combinations”, and Handbook Section 3062, “Goodwill and Other Intangible Assets”. Section 1581 requires that all business combinations be accounted for by the purchase method and it sets out criteria in determining the valuation and allocation of the purchase price in a business combination to tangible assets, intangible assets and goodwill. Section 3062 requires that goodwill no longer be amortized to earnings, but instead be periodically reviewed for impairment. Section 3062 also requires that intangible assets be assessed to determine if they have an estimated useful life or whether they have an indefinite life. Intangible assets that have an estimated useful life will continue to be amortized systematically over the useful life. Intangible assets with indefinite useful lives are not to be amortized but are instead to be tested for impairment annually.
Upon adoption of the new Section 3062 in fiscal 2003, the Company must perform transitional impairment tests on goodwill and intangible assets with indefinite lives. Any impairment losses are to be measured as of the date of adoption. Impairment losses assessed on transition, if any, will be recorded as an adjustment to retained earnings. The impact of adopting Section 1581 and 3062 has not yet been determined.
23
In July 2001, the U.S. Financial Accounting Standards Board (“USAcSB”) issued Statement of Financial Accounting Standard (“SFAS”) No. 141, “Business Combinations” and SFAS 142 “Goodwill and Other Intangible Assets” which are consistent with Sections 1581 and 3062, respectively, except for certain remaining generally accepted accounting principles differences, including the accounting for purchased in-process research and development and the recording of any impairment charge determined on transition as a period cost which are required under U.S. GAAP.
In December 2001, the AcSB issued Handbook Section 3870 “Stock-Based Compensation and Other Stock-Based Payments”. Section 3870 establishes standards for the recognition, measurement, and disclosure of stock-based compensation and other stock-based payments made in exchange for goods and services provided by employees and non-employees. It applies to transactions in which common shares, stock options, or other equity instruments are granted or liabilities incurred based on the price of common stock or other equity instruments.
The Company will adopt Section 3870 for its fiscal year beginning June 1, 2002. The Company does not believe that the adoption of this standard will have a material impact on the Company’s financial condition or results of operations as the Company’s current accounting policies, as disclosed above, comply with the new standard.
U.S. and Canada GAAP Difference
The Company’s financial statements have been prepared in accordance with generally accepted accounting principles as applied in Canada. In certain respects, GAAP as applied in the United States differs from that applied in Canada.
SFAS 123 Employee Stock Compensation
SFAS No. 123 encourages, but does not require, the recording of compensation costs for stock options issued to employees to be valued at fair value. For companies choosing not to adopt the fair value measurement for stock based compensation, the pronouncement requires the Company to disclose pro forma net income and earnings per share information as if the Company had accounted for its stock options under the fair value method since 1995. The Company has elected not to adopt the recording of compensation costs for stock options at fair value and, accordingly, a summary of the pro forma impact on the statement of loss is presented in the table below:
| (amounts in 000's) | 2002 | 2001 | 2000 | |||||||||
Loss for the year |
$ | 13,487 | $ | 15,213 | $ | 8,599 | ||||||
Compensation expense related to the
fair value of stock options |
1,278 | 1,059 | 1,285 | |||||||||
Pro forma loss for the period |
$ | 14,765 | $ | 16,272 | $ | 9,884 | ||||||
Pro forma loss per common share |
$ | 0.10 | $ | 0.12 | $ | 0.11 | ||||||
24
The fair value of each option granted has been estimated at the date of grant using the Black-Scholes option pricing model with the following assumptions used for options granted in the years ended May 31, 2002, 2001, and 2000: (i) dividend yield of 0%; (ii) expected volatility of 80% (2001 — 95%, 2000 — 95%); (iii) risk-free interest rate of 3.6% (2001 — 5.4%, 2000 — 6.0%) and (iv) expected lives of 5 years. The Company has assumed no forfeiture rate as adjustments for actual forfeitures are made in the year they occur. The weighted-average grant-date fair values of options issued in the years ended May 31, 2002, 2001, and 2000 were $ 0.71, $1.56, and $0.60 respectively.
SFAS 130 Reporting Comprehensive Income
SFAS No. 130 establishes standards for reporting and presentation of comprehensive income. This standard defines comprehensive income as the changes in equity of an enterprise except those resulting from shareholder transactions. Comprehensive loss for the periods presented in the Company’s financial statements equaled the loss for the period.
C. Research and Development, Patents and Licenses, etc.
See Note 8 to the Financial Statements at Item 17.
Cancer Therapy Technologies
Cancer Biotherapy
Chemotherapeutic drugs have been a major medical treatment for cancer for the past 30 years. However, a wide range of new cancer drugs have been developed by biotechnology companies that help improve patients’ quality of life. Unlike chemotherapies which are chemically based, these new drugs are biological, based on naturally occurring proteins or genetic material. The biotherapies in development include immunotherapy, gene therapy, and angiogenesis inhibitors. While chemotherapy drugs are typically toxic and delivered systemically, these biological agents are targeted to the tumor and, more specifically, to individual molecules or genes. These agents promise to have few and only mild side effects which means that, in theory, larger, and therefore more effective, doses can be administered.
The Company’s lead products span three classes of anti-cancer therapies: (i) immunotherapy, based on macrophage stimulating biologic response modifiers; (ii) antisense therapies, based on synthetic segments of DNA designed to bind to the messenger RNA (mRNA) that is responsible for the production of proteins over-expressed in cancer cells, and (iii) small molecule chemotherapies based on anti-angiogenic, anti-proliferative and anti-metastatic agents. The Company has a number of other anti-cancer technologies in the research and pre-clinical stages of development, including gene therapy and U-Sense technology.
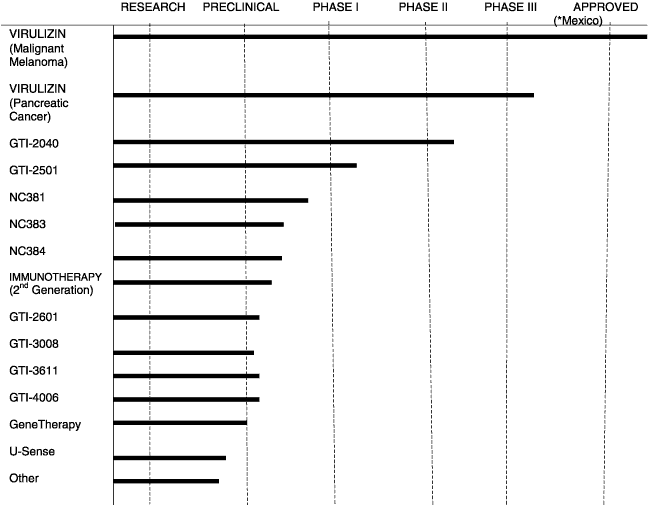
The Company’s product pipeline is illustrated below, summarizing the stage of development of the Company’s products.
25
PRODUCT PIPELINE
| * | Approved for sale in the private market in Mexico |
PRINCIPAL PRODUCTS
Immunotherapy
VIRULIZIN®
Immunotherapy is a form of treatment that stimulates the body’s immune system to fight diseases such as cancer. Immunotherapy may help the immune system to fight cancer by recognizing the difference between healthy cells and cancer cells, or it might stimulate the production of certain cancer fighting cells. VIRULIZIN® has been shown to be a non-toxic immunotherapy that recruits monocytes and macrophages to attack tumor cells. Since the drug works by encouraging the immune system to attack the cancer, rather than killing the cancerous cells itself, it can demonstrate fewer negative side effects than commonly used chemotherapy agents.
26
The human immune system and the body’s other protective cellular and molecular systems constitute a complex network of organs, tissues and cells which protect the body against foreign substances such as viruses, foreign tissue and cancer. Appropriate immune system response is critical to both health and survival. When the immune system functions properly, the system recognizes and effectively eliminates foreign substances. Conversely, inadequate or suppressed immune function may result in disease and, possibly, death. When inadequate or suppressed immune function occurs, modification or enhancement of the immune system may restore normal function. Immune system modification or enhancement may be achieved through the use of therapeutic products that stimulate or activate the immune system to achieve a desired response.
In recent years, a major focus of the biotechnology industry has been to develop naturally occurring human therapeutics, which are referred to broadly as biologic response modifiers (“BRMs”), and are so described because they are able to influence certain cellular events in the body. Many different substances are classified as BRMs and they have varied biological activities. Some of the major categories of BRMs include interferons (naturally occurring proteins capable of killing cancer cells or inhibiting their growth), interleukins (growth factors that stimulate cells of the immune system to fight cancer) and cytokines (substances produced by immune system cells, usually to send messages to other cells). BRMs have applications in a variety of diseases, including cancer, and are currently being employed in the area of cancer immunotherapy. BRMs may be used alone, in various combinations with other BRMs, or as adjuncts to other therapies.
VIRULIZIN®, the Company’s immunotherapeutic drug, has been shown to be a non-toxic immunotherapy that recruits monocytes and macrophages to attack tumor cells. Types of white blood cells, monocytes and macrophages are key players in the immune response to foreign invaders, including tumor cells. When macrophages and monocytes are activated, they produce proteins called cytokines, which have the ability to kill tumor cells directly. VIRULIZIN® stimulates the release of tumor necrosis factor (TNF-alpha), one type of cytokine, in immune cells to induce apoptosis (programmed cell death) of tumor cells. It is likely that the drug works by encouraging the immune system to attack the cancer, rather than killing the cancerous cells itself, studies suggest that VIRULIZIN® produces fewer negative side effects than commonly used chemotherapy agents.
Pre-clinical Testing
Toxicity studies conducted at independent laboratories have shown VIRULIZIN® to have a good safety profile. No demonstrable LD50 was determined during these studies, and repeated administrations of VIRULIZIN® did not result in organ system toxicities. In November 1998, additional preclinical data on the efficacy of VIRULIZIN® was obtained from studies performed at the University of Nebraska Medical Center. The Company performed these supporting studies to determine the efficacy of VIRULIZIN® in connection with gemcitabine, an Eli Lilly product that is the standard for first-line treatment of pancreatic cancer, in a human tumor xenograft model commonly used for pancreatic cancer. After extended daily administration, VIRULIZIN® significantly inhibited tumor growth in this model compared to a placebo. VIRULIZIN® also showed a trend towards an additive anti-tumor activity when combined with gemcitabine.
In January 2001, the Company reported that VIRULIZIN® demonstrated better anti-tumor activity in mice containing human breast cancer tumor cells than Taxol, one of the current standard treatments available for breast cancer. Additional findings revealed that the most outstanding anti-tumor activity occurred when VIRULIZIN® and Taxol were used in combination. Also, in August 2001, the Company announced pre-clinical results of VIRULIZIN® at the international conference, Drug Discovery Technology 2001. The results
27
of four independent tests with mice inoculated with human large cell lung carcinoma cells, small cell lung carcinoma cells, ovarian adenocarcinoma cells and prostatic carcinoma cells showed significant improvement over the current standard of treatment or the saline control.
Clinical Development Program
The clinical trials conducted by the Company were primarily established to determine the safety and efficacy of VIRULIZIN® as a single therapeutic agent for treating the most serious or life threatening cancer indications. These clinical trials involved Stage III and Stage IV cancer patients who had been diagnosed with cancers that were life-threatening and for which there were no established effective therapies. Approximately 250 patients had been enrolled in the clinical trials conducted in the United States, Canada, and Mexico and others have received VIRULIZIN® through the Company’s special access program.
The Company received orphan drug designation from the United States Food and Drug Administration (“FDA”) in February 2001 for VIRULIZIN® in the treatment of pancreatic cancer. Orphan drug status is awarded to drugs used in the treatment of a disease that afflicts less than 200,000 patients annually in the U.S. to encourage research and testing. This status means that the FDA will help to facilitate the drug’s development process by providing financial incentives and granting seven years of market exclusivity in the U.S. (independent of patent protection) upon approval of the drug in the United States.
The Company has initiated a Phase III clinical trial to evaluate VIRULIZIN® for the treatment of advanced pancreatic cancer. The Company intends to present the results of this clinical trial to the FDA in a new drug application at the completion of the study. This double-blind, randomized clinical trial is designed to be conducted at approximately 40 North American medical centres with the goal of enrolling 350 patients with advanced pancreatic cancer. Patients enrolled in the study will be randomised to receive either treatment with gemcitabine or treatment with gemcitabine in combination with VIRULIZIN®. Those patients who fail or become resistant to gemcitabine will then be treated with 5-Fluorouracil (5-FU) or with 5-FU in combination with VIRULIZIN®. The Company’s study protocol provides that all study subjects will be monitored throughout the remainder of their lifespan. The end points of the study will be survival and clinical benefits, and the duration is expected to be approximately four years. If the trial is successful, the Company intends to file an NDA with the FDA.
In June 2002, the Company announced that the FDA had granted “fast track designation” for VIRULIZIN® in the treatment of pancreatic cancer. This designation means that the FDA will assist in facilitating the development and expediting the review of VIRULIZIN®. Drugs given a fast track designation are intended for the treatment of a life-threatening condition and have demonstrated the potential to address unmet medical needs. Fast track products usually meet the FDA’s criteria for priority review.
28
Clinical Trial Results
In September 1992, the Company completed a Phase II clinical trial of VIRULIZIN® in Canada for the treatment of pancreatic adenocarcinoma. Pancreatic adenocarcinoma develops in the glands that produce enzymes that travel through the pancreatic duct to the small intestine to aid digestion. Approximately 90% of pancreatic cancers are pancreatic adenocarcinomas. The historic median survival from date of diagnosis for late stage pancreatic adenocarcinoma patients is approximately 120 days with a one-year survival rate of 13.8% (Brijir Gudjonsson, “Cancer of the Pancreas — 50 Years of Surgery” (1987) 60 Cancer 2284). In the Company’s Phase II clinical trial, the median survival for advanced pancreatic adenocarcinoma patients with an expectation of at least three months survival treated with VIRULIZIN® was 219 days, and the one-year survival rate was 35%. Disease stabilization was reported in 35% of the evaluable patients for more than three months. None of these patients developed any clinical or laboratory evidence of drug-related toxicity ((1994) 17 Clinical Investigative Magazine 37-41).
In September 1993, the Company completed a Phase II clinical trial of VIRULIZIN® in Mexico in the treatment of advanced malignant melanoma. Advanced malignant melanoma is a type of skin cancer with a tendency to spread via the lymphatic system and blood supply to other organs and tissues. The historic median survival from the date of diagnosis of advanced malignant melanoma was 93 days with a one-year survival rate of 13% (C.M. Balch, et al., eds., Cutaneous Melanoma, 2nd ed. (Philadelphia: J.B. Lippincott, 1992)). In the Company’s Phase II trial, the median survival from the date of diagnosis for advanced malignant melanoma patients treated with VIRULIZIN® was 396 days and the one-year survival rate was 54%. Only a few mild to moderate adverse events related to treatment with VIRULIZIN® were reported including pain at the injection site and fever. The interim results from this multi-center Phase II clinical trial were presented at the Baylor College of Medicine Research Symposium in April 1993. Based upon the results of the Mexican trial, the Company filed an NDA in November 1996, to obtain marketing approval of VIRULIZIN® in Mexico as a treatment for advanced malignant melanoma. In October 1997, the Company received a license from the SSA to sell VIRULIZIN® in Mexico in the private market for the treatment of malignant melanoma.
In August 1998, the Company released results of the Phase I/II trial evaluating VIRULIZIN® in patients with pancreatic cancer at the Rush Cancer Institute. Of the 26 patients enrolled, 19 were deemed evaluable according to the study protocol. The Company announced that the overall median survival for all evaluable patients was 6.7 months and the six-month survival rate was 58%. These results confirm and extend previous studies performed by the Company in Canada in pancreatic cancer patients. Results of the current study have also shown that VIRULIZIN® continues to show an excellent safety profile, and there is an increasing trend and a statistically significant improvement in total quality-of-life change score.
The first peer-reviewed presentation of the U.S. preclinical and clinical trial results of VIRULIZIN® was presented at the 90th Annual Meeting of the American Association for Cancer Research (the “AACR”) in Philadelphia on April 13, 1999. A scientific abstract, entitled “A Novel Immunotherapeutic Agent for Pancreatic Cancer: Results of Preclinical and Clinical Trials Using VIRULIZIN®” was submitted to the AACR in October 1998. During the AACR meeting, it was announced that in a preclinical study in mouse models of human pancreatic cancer, VIRULIZIN® significantly inhibited tumor growth. Among treated mice, tumors were only 60% of the size of the tumors in the untreated mice. The response was even greater when the chemotherapeutic agent gemcitabine was added to the treatment, with tumors in the treated mice only 26% of the size of the tumors in the untreated mice.
29
Future Applications
The Company believes that in vitro and in vivo research supports the therapeutic potential of VIRULIZIN® in the treatment of diseases associated with immune system disorders other than cancer. The Company previously sponsored research studies at Rush-Presbyterian-St. Luke’s Medical Center in Chicago to study the potential of VIRULIZIN® in the treatment of endometriosis. The Company’s scientists have also conducted pre-clinical research in the use of VIRULIZIN® in combination with known cytotoxic or chemotherapeutic agents in the treatment of cancer. The Company intends that the results from these studies will form the basis for a potential clinical program of VIRULIZIN® in combination with other cancer therapeutic agents. However, there can be no assurance that the Company will enter into this clinical program.
In April 2002, the Company presented data supporting both the mechanism of action and the characterization of VIRULIZIN®, at the American Association for Cancer Research Conference. This knowledge of the composition of VIRULIZIN® has enabled the Company to initiate a pre-clinical research program for the development of novel immunotherapeutic products. One such product has recently been tested in vivo and compared to VIRULIZIN® for anticancer activity. Results from these pre-clinical tests indicated that VIRULIZIN® and the new immunotherapeutic product exhibited strong antitumor activity in mouse models with human pancreatic carcinoma tumors.
Antisense therapies
Many chemotherapeutic drugs are chemicals designed to induce or inhibit the function of a target molecule, typically an enzyme or receptor. The selectivity of these drugs is usually determined by only a few, generally two or three, points of interaction at the binding site of a target molecule. Frequently, sites on other non-target molecules resemble the target-binding site sufficiently to permit the conventional drug to bind to some degree. This indiscriminate affinity or lack of specificity can result in decreased efficacy, unwanted side effects, and increased toxicity. Overcoming these limitations has been a primary goal for recent anti-cancer drug development. One such method involves the use of antisense therapies.
The human metabolism is essentially controlled by proteins produced by the body. Since most human diseases, including cancer, can be traced to faulty protein production, traditional therapeutics are designed to interact with the disease causing proteins. Antisense therapeutics take a different approach to treatment: they are designed to prevent the production of the proteins causing the disease.
Antisense oligonucleotides are synthetic segments of DNA designed to bind to the mRNA that is responsible for the production of disease-associated proteins. The sequence of nucleic acid bases in an antisense oligonucleotide is complementary to its nucleic acid target sequence on the mRNA. Thus, the antisense oligonucleotide binds at a significant number of points to the target site, and hybridizes (binds) tightly to the selected mRNA. Since a single mRNA may be translated repeatedly into a protein, a single antisense oligonucleotide may inhibit the synthesis of many copies of a protein. Moreover, in vitro tests have shown that these antisense/mRNA complexes activate enzyme activity that destroys the mRNA to which the oligonucleotide is bound, without destroying the oligonucleotide itself. This frees the oligonucleotide to bind with another identical target mRNA and repeat the inhibitory process. Furthermore, it is possible that an antisense molecule may inhibit specific expression by binding to the gene responsible for coding for the mRNA molecule preventing the mRNA molecule from being synthesized. It may also bind to an unprocessed mRNA
30
molecule, preventing it from developing into a mature mRNA, or it may bind to a specific mRNA molecule preventing it from translocating from the nucleus of the cell to the cytoplasm where protein synthesis occurs.
The main attributes of antisense therapies are specificity and rational design. As a function of the simple nucleotide base-pairing rules, therapeutic intervention using antisense compounds can be a universal approach to a number of diseases whose causative agents or targets have been characterized at the DNA level. The specificity of an antisense therapeutic is determined by the occurrence of a given nucleotide sequence. It has been calculated that a 17-mer (17 nucleotides) oligonucleotide sequence should theoretically appear only once in the human genome. Furthermore, the binding affinity between complementary strands of nucleic acids is exceptionally high. It is expected that antisense’s combination of high specificity and affinity, which is difficult to achieve with conventional protein-targeted drugs, could substantially reduce side effects due to unwanted interference with other essential cellular activities.
The rational design of oligonucleotides aimed at the inhibition of gene expression is based on targeting nucleotide sequences. Thus, an advantage of developing antisense compounds is that the design and synthesis are relatively straightforward. The structure of 20-mer antisense oligonucleotides, which is complementary to all possible 20 base sequences within a 1,000 base mRNA sequence, is strictly defined.
The unique premise of this therapeutic approach is to target an earlier stage of the biochemical process than is usually possible with conventional drugs. Traditional therapies usually interact with the final synthesized or processed protein, whereas this newer approach alters an earlier expression of the gene that codes for such a protein. Therefore, it is believed that drugs based on this approach may have broad applicability, greater efficacy and fewer side effects than conventional drugs.
The effectiveness of an antisense drug is largely dependent on the protein targeted. When examining antisense targets, the Company chose ribonucleotide reductase (RNR) because of its significance in the uncontrolled cell growth associated with essentially every cancer. RNR, which is composed of two protein components called R1 and R2, is a highly regulated cell enzyme that is essential for DNA synthesis and repair. RNR catalyzes the formation of deoxyribonucleotides, which are required for building the cell’s DNA and thus responsible for cell replication. The Company’s antisense therapeutics are designed to inhibit this process, and in turn, the Company anticipates that this will inhibit the growth of tumor cells.
GTI-2040
The Company’s lead antisense therapy is GTI-2040. GTI-2040 is an antisense agent that targets the R2 component of RNR. The R2 component is an unusually effective target for drug intervention because it is the rate-limiting component in the ribonucleotide reductase activity important for cancer cell proliferation. It is also bi-functional since an elevation in the R2 component in cancer cells also alters the activity of an important biochemical signal pathway called the MAP Kinase Pathway, which is known to play a role in mechanisms leading to the development of cancer. The Company has designed antisense molecules that specifically target the R2 mRNA, resulting in:
| • | reduced R2 protein and R2 mRNA levels in human tumor cells grown in culture; | ||
| • | significant inhibition of tumor cell growth in vitro; and | ||
| • | statistically significant reduction of tumor growth and tumor cell dissemination (metastasis) in animal models. |
31
Since it has been noted that levels of R2 are elevated in cancer cells, an antisense molecule that binds to the mRNA coding for R2 inhibits the replication of diseased cells. Further, research indicates that reducing levels of R2 expressed in cells lowers resistance levels to other pharmaceutical compounds that might be used in combination therapy with GTI-2040.
GTI-2501
The Company’s other antisense therapy is GTI-2501, designed to specifically target the R1 mRNA, resulting in:
| • | reduced R1 protein and R1 mRNA levels; | ||
| • | significant inhibition of tumor cell growth in vitro; | ||
| • | statistically significant reduction of tumor growth and tumor cell dissemination (metastasis) in animal models; and | ||
| • | total regression in some tumor models. |
GTI-2501 is an antisense agent that targets the R1 component of human RNR and research indicates that it prevents the formation of the R1 protein required for ribonucleotide reductase activity.
Pre-Clinical Testing
Pre-clinical studies have demonstrated that both GTI-2040 and GTI-2501 are well tolerated in humans at concentrations that exceed therapeutic doses. All observed toxicities are consistent with phosphorothioated oligonucleotide compounds, and include increased blood clotting times as well as some signs of complement activation at high doses.
In the presence of GTI-2040, both R2 protein and R2 mRNA levels in tumor cells in vitro were dramatically reduced. Experiments have demonstrated that GTI-2040 specifically alters the expression of its target, R2, in cancer cells. Tumor suppression in a widely accepted mouse model has been demonstrated in a variety of tumor types, including colon, pancreas, liver, lung, breast, ovarian, brain, kidney and skin. The effect on tumor suppression is statistically significant in all cases, with significance at p [less than or equal to] 0.0001 for colon, pancreas, kidney and lung cancers. These results suggest that GTI-2040 has the potential to be a strong drug candidate for the treatment of a broad range of human cancer types.
On November 22, 1999, the Company announced results of anti-tumor studies with GTI-2040, including complete tumor regressions, when used in combination with certain chemotherapeutic agents. The results were based on in vivo studies performed using recognized human kidney cancer, colon cancer and melanoma models. The studies were performed at the Company’s research facilities at the Sunnybrook Health Sciences Centre in Toronto, Canada and have been confirmed through repeat experiments.
The Company reported that when SCID mice bearing a human kidney cancer line were treated with a combination of GTI-2040 and two commonly used chemotherapy drugs (5-FU or vinblastine), complete tumor regression was observed in all the mice treated. The Company also reported that similar tumor regressions were observed in studies combining GTI-2040 and another commonly used chemotherapy drug, mitomycin C, in in vivo mouse models of human melanoma and human colon cancer. GTI-2040 also significantly enhanced the anti-tumor effects of other well-known anti-cancer drugs such as gemcitabine, estramustine and paclitaxel in a standard human colon carcinoma mouse model.
32
In July 2001 the Company announced that GTI-2040 demonstrated anti-tumor activity in animal models with human lymphoma tumors, adding to the already broad range of cancers in which GTI-2040 has shown anti-tumor activity.
GTI-2501 targets R1 and has been selected as the Company’s second antisense drug candidate. Complete tumor regression was seen in mice bearing human breast and renal derived tumors. These findings have been reproduced by both academic laboratories and interested partners. The compound was also found to be highly active against a broad range of other human tumors in these animal models. On November 29, 1999, the Company reported that GTI-2501 was an effective anti-cancer agent when tested in standard mouse models bearing a variety of different human cancer lines including tumor cells derived from lung, breast, colon, kidney, ovary, pancreas and skin cancer. The greatest anti-tumor activity was found with human tumor cells derived from kidney and breast cancers treated with GTI-2501. In three independent tests using two different human kidney cancer lines, the Company observed complete tumor regressions in all 20 mice tested.
The anti-tumor activity of both GTI-2040 and GTI-2501 was further demonstrated in mouse models containing human prostate cancer cells. In February 2000, the Company observed that while both drugs produced marked reductions in tumor growth, the GTI-2501 treatment also led to disease stabilization (i.e. very little or no tumor growth).
Clinical Development Program
Formal preclinical development of GTI-2040, including manufacturing and toxicology studies, was initiated in mid-1998. An IND application filed with the FDA was approved on December 7, 1999 for a Phase I/II clinical trial of GTI-2040, given as a 21-day continuous intravenous infusion in the treatment of solid tumor and lymphoma. This trial was initiated in December 1999 under the direction of Dr. Richard Schilsky of the Chicago Cancer Research Center. 30 patients with advanced or metastatic solid tumors were enrolled in this study. Phase I clinical endpoints for safety and tolerability were met. Doses ranging from 18.5 mg/m2/day to 222.0 mg/m2/day have been studied and found to display favourable safety profiles. Based on studies indicating that the R2 target would be down-regulated at concentrations of GTI-2040 of 185.0 mg/m2/day, the recommended Phase II dose for GTI-2040 administered as a single agent was 185.0 mg/m2/day. An additional six patients with renal cell carcinoma were enrolled in this study to further characterize the toxicity in this patient population.
The Company has a Phase II program underway for GTI-2040. This program includes an open-label, non-randomized study of GTI-2040 in combination with capecitabine in patients with advanced metastatic renal cell carcinoma. This trial is being conducted by Dr. Frank Torti at Wake Forest University in North Carolina. The decision to select this drug combination (GTI-2040 and capecitabine) was based on pre-clinical and clinical studies that indicated that GTI-2040 or the combination of GTI-2040 and 5-FU, the active ingredient in capecitabine, should provide benefits for patients with renal cell carcinoma.
In June 2002, the Company announced that it has been approved by the Drug Development Group of the Division of Cancer Treatment and Diagnosis, the National Cancer Institute (NCI), to supply the Company’s lead antisense drug, GTI-2040, for multiple clinical trials to evaluate its efficacy in a range of cancers. The NCI approved GTI-2040 after an analysis of pre-clinical, good laboratory practice toxicology and Phase I clinical data. The Company intends to work together with the NCI to select cancer indications and suitable development programs for a number of clinical trials. The NCI has also indicated it will sponsor trials conducted with GTI-2040 alone or in combination with other cancer therapies.
33
GLP-toxicology studies for GTI-2501 were completed in November 2000, and approval of an IND was received from the U.S. FDA February 2001. This Phase I dose-escalating study is underway at the University of Chicago Medical Centre and is designed to establish the recommended clinical Phase II dose as well as look at the safety profile of GTI-2501. Patients with solid tumors or lymphoma are being enrolled.
Small Molecule Chemotherapies
Most currently employed anti-cancer chemotherapeutic drugs are DNA damaging, cytotoxic agents, designed to act on rapidly dividing cells. These drugs typically include unpleasant or even serious side-effects due to their non-specificity to cancer cells, and frequently lead to tumor-acquired drug resistance. As a result of these limitations, there is an ongoing intensive world-wide effort to discover more effective anti-cancer drugs.
In December 1997, the Company, through NuChem, acquired certain patent rights and a sublicense from Ion to develop and commercialize the anti-cancer applications of CLT and new chemical entities related to CLT (the “NuChem Analogues”). The consideration for this acquisition was Ion’s 20% common share interest in NuChem, US$350,000 in common shares of the Company and amounts totalling up to US$3,500,000 payable in cash. On June 15, 1998, the Company issued from treasury 583,188 common shares in settlement of the US$350,000 obligation. To August 31, 1999, NuChem had made cash payments totalling $714,750 (US$500,000) to Ion. The balance is payable upon the achievement of certain milestones based on the commencement and completion of clinical trials related to the NuChem Analogues.
All research and development activities to be undertaken by NuChem are to be funded by the Company through subscriptions for non-participating preference shares of NuChem. As at May 31, 2002, the Company had provided a total of $5,983,000 of funding to NuChem.
NuChem has agreed to actively proceed with research and development programs relating to the NuChem Analogues. If NuChem fails to make any of the payments described above, discontinues its research and development activities related to the NuChem Analogues or commits an act of insolvency or bankruptcy, Ion has the right to re-acquire the NuChem Analogues assigned to NuChem and to terminate the sublicense, upon the payment by Ion of a certain amount and subject to certain other conditions.
The Company and Ion are parties to a unanimous shareholders’ agreement relating to NuChem. Under that agreement, the Company has the right to appoint NuChem’s officers and a majority of the members of NuChem’s board of directors. The Company directs the business and operations of NuChem, subject to the terms of an annual business plan that Ion is entitled to approve. Any profits that are distributed from NuChem will be shared between the Company and Ion in proportion to their ownership of NuChem common shares. The unanimous shareholders’ agreement provides mutual restrictions on the transfer of NuChem shares, as well as mutual rights of first refusal, drag-along and piggyback rights. If Ion becomes entitled to re-acquire the NuChem Analogues assigned to NuChem and to terminate the sublicense, the Company is entitled to purchase Ion’s NuChem shares for their stated capital amount.
Pre-clinical and Investigational Studies
Through NuChem, the Company is currently pursuing research and pre-clinical development of NuChem Analogues for the treatment of a wide variety of cancers.
34
In March 1998, the Company signed an agreement with the Developmental Therapeutics Branch, Division of Cancer Treatment of NCI, under which that organization will screen anti-cancer compounds in its panel of cancerous cell lines. The data from these tests was used, in part, as the basis for selecting the compounds which will be given priority for entering clinical trials. The Company also signed an agreement with Harvard Medical School for the conduct of various research projects related to the further development of CLT and the NuChem Analogues as anti-cancer agents.
On February 9, 1999 the Company announced, on behalf of NuChem, additional positive preclinical efficacy data on four of NuChem’s novel anti-cancer compounds. This in vivo study performed by Dr. José Halperin at Harvard Medical School involved a murine (mouse) KLN squamous cell cancer prevention model. The goal of the study was to specifically assess the efficacy of the analogues upon oral administration. Relative to controls, this efficacy study showed significant inhibition of squamous cell carcinoma tumors by the test compounds and no apparent toxic effect due to the treatment regimen was noted in the animals. The results confirm the efficacy results obtained in an earlier study for several of NuChem’s novel anti-cancer compounds.
Agreements were concluded in 1998 and 1999 with other institutions and contract research organizations for the conduct of studies on certain NuChem Analogues to support the selection of one or more lead compounds for pre-clinical development toward an IND submission. The University of Nebraska Medical Center (Omaha, Nebraska) conducted studies of pre-clinical toxicity in normal mice and efficacy studies in human cancer xenograft (mouse) models. Phoenix International Life Sciences Inc. (Montréal, Québec) conducted studies to measure CLT analogues in biological matrices by a LC-MS/MS method, and to determine the metabolic stability and metabolite profile of certain CLT analogues by in vitro methods. McMaster University (Professor Jack Rosenfeld) conducted bioanalytical method development for the measurement of CLT analogues in plasma and tissues of mice by HPLC.
On February 1, 1999, positive preclinical data on the efficacy of the Company’s anti-cancer compounds were obtained from a second study performed at the University of Nebraska Medical Center (Omaha, Nebraska). This study was designed to confirm the efficacy of certain novel anti-cancer (CLT Analogues) compounds in a human tumor xenograft model commonly used for colon cancer. The Company announced on behalf of NuChem independent confirmation by NCI of the in vitro anti-cancer activity of its novel compounds currently under development. Later in 1999, the Company announced on behalf of NuChem that the NCI had agreed to allocate resources for the further development of three novel anti-cancer compounds discovered at Harvard Medical School to investigate formulation development and the pharmacology of the compounds.
On February 7, 2000, the Company announced that NuChem had determined that one of its key anti-cancer CLT analogues, NC381, showed positive pre-clinical results in inhibiting the spread of human melanoma tumor cells in mice. NC381, whether administered through injection or orally, exhibited anti-metastatic activity with few apparent side effects on 16 mice.
NC381 has demonstrated pre-clinical activity in lung, pancreatic and kidney cancers as well as anti-angiogenic, anti-proliferative, and anti-metastatic properties. Research suggests that it can be taken orally, which positions it as a potential maintenance therapy. NC381 requires further testing to complete its ongoing pre-clinical development in Canada and the United States before the Company can file an IND to obtain approval to initiate human clinical trials either in Canada or the United States as a treatment for various forms of cancer. However, there can be no assurance that such approval will be obtained on a timely basis, if ever. See “Business of the Company — Regulatory Strategy”.
35
Other Technologies
Several promising new product opportunities have been introduced to the Company’s portfolio and are being assessed for their potential as new drug candidates. They include platform technologies in areas of tumor suppressor gene therapy, and U-Sense compounds that the Company believes to have the potential to work through a unique mechanism of action to decrease the expression of cancer relevant genes. Further antisense approaches for the treatment of cancer and drug resistant bacteria are also being investigated in the Company’s laboratory. The Company intends to continue developing these compounds with the aim to identify new drug candidates for clinical trials as the three lead drugs make their way through clinical trials and to market.
D. Trend Information
Lorus has not produced or commercially marketed any product. Although the Company has entered into an agreement in respect of the commercial sale of Virulizin® in Mexico, there can be no assurance that any sales of the product will be made. The Company’s future profitability is dependent on the successful outcome of clinical trials and the subsequent successful marketing and/or partnering of its products.
The Company does not expect to generate a positive cash flow from operations for several years due to substantial additional research and development costs, including costs related to drug discovery, preclinical testing, clinical trials, manufacturing costs and operating expenses associated with supporting these activities. The Company may need to raise additional capital to fund operations over the long-term.
36
Item 6. Directors, Senior Management and Employees
A. Directors and Senior Management
The following table and notes thereto provide the name, municipality of residence, positions with the Company, term of office and principal occupation of each person who serves as a director or officer of the Company as at November 30, 2002. Officers serve at the discretion of the Board of Directors.
Each director has been elected or appointed to serve until the next annual meeting or until a successor is elected or appointed. The Company has an Audit Committee, an Environmental Committee, a Corporate Governance Committee and a Human Resources and Compensation Committee and the members of each such committee are shown below. As at December 6, 2002, the directors and executive officers of the Company, as a group, beneficially owned, directly or indirectly, or exercised control over 14,292,880 or approximately 10% of the common shares.
| Name and Municipality | Director or | |||||||
| of Residence | Position | Officer Since | Age | |||||
| ROBERT BÉCHARD(1)(2) Montreal, Quebec |
Director | October 1999 | 44 | |||||
| SUZANNE CADDEN Mississauga, Ontario |
Vice President, Clinical and Regulatory Affairs | July 2001 | 44 | |||||
| GEOFFREY COLLETT Mississauga, Ontario |
Vice President, Corporate Development | March 2000 | 49 | |||||
| SHANE A. ELLIS Toronto, Ontario |
Corporate Secretary, Vice President of Legal Affairs | April 1998 | 49 | |||||
| JAMES T. PARSONS Mississauga, Ontario |
Vice President, Finance and Administration and Chief Financial Officer | March 2000 | 37 | |||||
| DONALD W. PATERSON(1) Toronto, Ontario |
Director | July 1991 | 70 | |||||
| ELLY REISMAN Toronto, Ontario |
Director | November 1999 | 52 | |||||
| ALAN STEIGROD(2) Newport Beach, California |
Director | May 2001 | 65 | |||||
| GRAHAM STRACHAN(1)(3)(4) Etobicoke, Ontario |
Chair of the Board of Directors, Director | May 2001 | 64 | |||||
| DR. JIM WRIGHT Oakville, Ontario |
Chief Executive Officer, Director | October 1999 | 60 | |||||
| DR. AIPING YOUNG(4) Toronto, Ontario |
Senior Vice President, Research and Development and Chief Technology Officer | October 1999 | 46 | |||||
| (1) | Member of Audit Committee. | |
| (2) | Member of the Human Resources and Compensation Committee. | |
| (3) | Member of the Corporate Governance Committee | |
| (4) | Member of Environmental Committee. |
37
The principal occupation and employment of each of the foregoing persons for the past five years is set forth below:
Robert Béchard: Mr. Béchard is currently a partner of RBC Capital Partners. From 1991 to 1996, Mr. Béchard was an Account Manager with a Canadian chartered bank and from 1996 to 1998, was an Investment Manager of Sofinov Société Financière D’Innovation Inc.
Suzanne Cadden: Ms. Cadden joined the Company in February 2001 as Director, Regulatory Affairs and Compliance. Prior to joining Lorus, Ms. Cadden was a Senior Director and Director of Regulatory Affairs and Compliance with Glaxo Wellcome Canada from 1996 to 2000. Prior to August 1996, Ms. Cadden was a Director of Regulatory Affairs and Pharmacoeconomics with CIGA-Geigy Canada.
Geoffrey Collett: Prior to joining the Company in March 2000, Mr. Collett was a Vice President within MDS Capital Corp., from October 1997 to March 2000. From 1988 to 1997, Mr. Collett worked with GlaxoWellcome as Licensing Manager and Director of Business Development and Planning.
Shane A. Ellis: From 1994 to 1997, Mr. Ellis lectured in business law at the School of Business Management, Ryerson Polytechnical University. From 1996 to 1998, Mr. Ellis acted as counsel for the Bennett & Wright Group of Companies.
James T. Parsons: Prior to joining the Company in March 2000, Mr. Parsons was Controller and Assistant Treasurer for Timminco Limited. Prior to November 1998, Mr. Parsons was Controller for Timminco.
Donald W. Paterson: Mr. Paterson is President of Cavandale Corporation, a company principally engaged in providing strategic corporate consulting to emerging growth companies within the technology industry. Prior to founding Cavandale Corporation, Mr. Paterson was a Director and Vice-President of Wood Gundy Inc., a Canadian investment bank, where he was directly involved in leading the firm’s activities in financing Canadian and international high technology companies.
Elly Reisman: Mr. Reisman is the President and Chief Executive Officer of Great Gulf Group, a real estate company. Mr. Reisman has held that position for more than the last five years.
Alan Steigrod: Mr. Steigrod is Managing Director of Newport Healthcare Ventures, a consulting firm for the healthcare industry, located in Newport Beach, California. Mr. Steigrod has held that position for more than the last five years.
Graham Strachan: Mr. Strachan is President of GLS Business Development Inc. a life-science consulting firm, located in Etobicoke, Ontario. Prior to 1999, Mr. Strachan was President, Chief Executive Officer and Director of Allelix Biopharmaceuticals Inc.
Dr. Jim Wright: Dr. Wright’s present principal occupation is Chief Executive Officer of the Company. He is also a member of the Lorus Board of Directors. Prior to October 1, 2001, Dr. Wright was President of the Company, a position he held since October 29, 1999. Prior to October 29, 1999, Dr. Wright was President and Chief Scientific Officer of GeneSense and a member of the board of GeneSense. He also served as Chairman of the Board. Prior to September 1998, Dr. Wright was Professor of Microbiology, Professor of Biochemistry and Molecular Biology, and Adjunct Professor of Internal Medicine at the University of Manitoba, Senior Scientist and Associate Director of the Manitoba Institute of Cell Biology and Terry Fox Senior Scientist of the National Cancer Institute of Canada.
38
Dr. Aiping Young: Prior to June 1996, Dr. Young was Senior Scientist, Group Leader and Medical and Scientific Advisor for Pias Corporation in Japan. From 1996 to 1999, Dr. Young was Vice President of Research and Development, and a member of the Board of Directors for GeneSense Technologies Inc. She has also been an Adjunct Scientist at the Manitoba Institute of Cell Biology at the Manitoba Cancer Foundation.
Medical and Scientific Advisory Board
On October 2, 2002 the Company announced the appointment of Mace L. Rothenberg M.D., as its external medical advisor providing strategic medical advice on the Company’s growing international clinical and drug development programs. Dr. Rothenberg is an internationally recognized oncologist. His research focuses on the evaluation of the effects of new drugs in humans from clinical pharmacologic, biologic and genetic perspectives. Dr. Rothenberg is an Ingram Professor of Cancer Research at the Vanderbilt-Ingram Cancer Center as well as Professor of Medicine at the Vanderbilt University Medical Center and a Direct of Drug Development.
The Company has a Medical and Scientific Advisory Board (“MSAB”) comprised of certain medical and scientific experts whom the Company believes will enhance its capabilities. Members of the MSAB meet periodically to review the progress of the Company’s research and development activities and the results of ongoing clinical trials. The MSAB also advises the Company generally as to specific research programs, and as to advances in biotechnology, immunology and other areas of scientific expertise relevant to the further development of the Company’s technologies.
As at December 6, 2002, the members of the MSAB were:
Dr. Donald P. Braun, Ph.D.: Dr. Braun is a Professor, Department of Surgery at the Medical College of Ohio in Toledo, Ohio, and Administrative Director of the Cancer Institute. Dr. Braun is a member of the Scientific Advisory Committee on Immunology of the American Cancer Society and is Chair of the Company’s MSAB.
Dr. Gregory Curt, M.D.: Dr. Curt is a Clinical Director of the NCI in Bethesda, Maryland. He received his M.D. with Distinction in Research from the University of Rochester School of Medicine in 1977. After completing his training in Internal Medicine at Harvard, he came to the NCI for subspecialty training in Medical Oncology.
Dr. Jaime G. de la Garza Salazar, M.D.: Dr. de la Garza, a member of the Mayo Graduate School of Medicine, has been the Director General of the National Cancer Institute of Mexico since 1993. He is also the President of the Mexican Oncology Board. Prior to his appointment as Director, Dr. de la Garza was Associate Director in Clinical Research at the NCI.
Dr. Robert Kerbel, Ph.D.: Dr. Kerbel obtained his Ph.D. in microbiology and immunology from Queen’s University in 1972. Dr. Kerbel is currently the Director of Biological Sciences and the Division of Cancer Biology Research at the Sunnybrook Health Science Centre in Toronto and is also the John & Elizabeth Tory Professor of Experimental Oncology at the University of Toronto. He is a member of the editorial board of many international scientific journals and is editor-in-chief of Cancer Metastatis Review.
39
Dr. Bishnu D. Sanwal, Ph.D., D.Sc., F.R.S.C.: Dr. Sanwal is a Professor Emeritus and former Chairman of the Department of Biochemistry at the University of Western Ontario, London, Ontario. He has a long and distinguished career in biological and medical research. With over 146 publications, Dr. Sanwal is a member of or advisor to numerous scientific committees and journals such as the editorial board of Archives of Biochemistry and Biophysics and member of the Royal Society of London, and Fellow of the Royal Society of Canada. He received a Ph.D. from the University of Delhi and a Doctor of Sciences from the Federal Institute of Technology, Zurich.
Dr. Lesley Seymour, MBBCh, FCP (SA): Dr. Seymour is a Co-Director of the Investigational New Drug Program of the National Cancer Institute of Canada Clinical Trials Group. She received her M.D. at the University of the Witwatersrand in South Africa in 1978 and subsequently completed Specialist training in Internal Medicine as well as Clinical Hematology and Medical Oncology.
Dr. Louis Siminovitch, O.C., Ph.D., D.Sc., F.R.S.C., F.R.S.: Dr. Siminovitch is a former founder and director of the Department of Medical Genetics, University of Toronto, the Department of Genetics, Hospital for Sick Children, the Samuel Lunenfeld Research Institute at Mount Sinai Hospital, former Director and President of the National Cancer Institute of Canada and is presently on the Scientific Advisory Board of the Canadian Medical Discoveries Fund, several biotechnology companies and institutes. He is a founding member and former Senior Editor of Virology, founding member and former member of the editorial board of Cell, the editorial board of Annual Review of Genetics, founding member and former Senior Editor of Molecular and Cellular Biology, former member of the editorial board of Genetics and of the advisory board of Molecular Biology and Medicine. Dr. Siminovitch received a Ph.D. from McGill University and was awarded a Doctor of Science, Honoris Causa, for his distinguished scientific research contributions from several Canadian universities: Memorial University, McMaster University, University of Montreal, McGill University, University of Western Ontario and University of Toronto. Dr. Siminovitch is a Companion of the Order of Canada and was inducted into the Canadian Medical Hall of Fame in 1997.
Dr. George R. Stark, Ph.D., F.R.S.: Dr. Stark is the Sherwin-Page Chairman of the Research Institute, The Cleveland Clinic Foundation, Cleveland, Ohio. He received a Ph.D. from Columbia University and completed postdoctoral studies at Rockefeller University. Dr. Stark has made significant contributions to the field of molecular biology. Dr. Stark led the development of the Northern and Western Blot techniques for analysis of specific RNAs and proteins. Much of his work has focused on the process of gene amplification in mammalian cells, leading to an appreciation both of the mechanisms that generate amplified structures in cell lines and tumor cells and the regulatory processes that prevent amplification from occurring in normal cells. Very recent work has led to the discovery of a new signal pathway that regulates gene expression in cancer cells. A former Professor of Biochemistry at Stanford University, Dr. Stark moved to London as the Associate Director of Research at the Imperial Cancer Research Fund in London, England (1983-1992). Dr. Stark received the H. A. Sober award of the American Society of Biological Chemists in 1986, was elected to the U.S.A. National Academy of Science in 1986 and to the Fellowship of the Royal Society in Britain in 1990.
B. Executive Compensation
Summary Compensation
The following Summary Compensation Table, presented in accordance with the regulation to the Securities Act (Ontario), sets forth the compensation paid in respect of individuals who were, in respect of the financial year ended May 31, 2002, the Chief Executive Officer and the four other most highly compensated executive officers of the Company (collectively, the “Named Executives”).
40
Summary Compensation Table
| Long-Term | ||||||||||||||||||||||||
| Compensation | ||||||||||||||||||||||||
| Awards | ||||||||||||||||||||||||
| Annual Compensation | Securities | |||||||||||||||||||||||
| Under | ||||||||||||||||||||||||
| Other Annual | Options/SARs | |||||||||||||||||||||||
| Salary | Bonus | Compensation | Granted | All Other | ||||||||||||||||||||
| Name and Principal Position | Year | ($) | ($) | ($) | (#) | Compensation | ||||||||||||||||||
Dr. Jim A. Wright(1) |
2002 | 274,885 | 61,480 | Nil | 300,000 | Nil | ||||||||||||||||||
Chief Executive Officer |
2001 | 196,231 | 65,675 | Nil | 150,000 | Nil | ||||||||||||||||||
| 2000 | 109,237 | 43,750 | Nil | Nil | Nil | |||||||||||||||||||
Dr. Raafat Fahim(2) |
2002 | 150,831 | Nil | Nil | 1,500,000 | 463,669 | ||||||||||||||||||
Former President and Chief |
2001 | Nil | Nil | Nil | Nil | Nil | ||||||||||||||||||
Operating Officer |
2000 | Nil | Nil | Nil | Nil | Nil | ||||||||||||||||||
Dr. Aiping Young(1) |
2002 | 190,154 | 24,480 | Nil | 200,000 | Nil | ||||||||||||||||||
Senior Vice President, |
2001 | 167,538 | 30,030 | Nil | 163,297 | Nil | ||||||||||||||||||
Research and Development |
2000 | 94,654 | 36,075 | Nil | Nil | Nil | ||||||||||||||||||
Mr. Geoffrey Collett(3) |
2002 | 165,846 | 20,073 | Nil | 225,000 | Nil | ||||||||||||||||||
Vice President, Corporate |
2001 | 152,000 | 24,850 | Nil | 50,000 | Nil | ||||||||||||||||||
Development |
2000 | 29,167 | 6,795 | Nil | 100,000 | 20,000 | ||||||||||||||||||
Mr. James T. Parsons(3) |
2002 | 155,846 | 19,865 | Nil | 100,000 | Nil | ||||||||||||||||||
Vice President, Finance |
2001 | 142,000 | 23,075 | Nil | 50,000 | Nil | ||||||||||||||||||
and Administration and CFO |
2000 | 30,038 | 6,310 | Nil | 120,000 | Nil | ||||||||||||||||||
| (1) | Dr. Wright and Dr. Young joined the Company on October 29, 1999, the date of acquisition of GeneSense Technologies Inc. by the Company. | |
| (2) | Dr. Raafat Fahim resigned from his position as President and Chief Operating Officer on July 31, 2002. The amount of “All Other Compensation” relates to June and July, 2002 compensation, a signing bonus at the time of hire, and a lump sum payment agreed with Dr. Fahim. | |
| (3) | Mr. Collett and Mr. Parsons joined the Company in March 2000. |
Executive officers of the Company, including the Named Executives (collectively the “Executive Officers”), are eligible to participate in a performance related compensation plan (the “Compensation Plan”). The Compensation Plan provides for potential annual cash bonus payments and annual granting of options to purchase common shares under the Company’s stock option plan. The potential annual cash bonus and annual granting of options to each Executive Officer are conditional upon the achievement by the Company and each Executive Officer of predetermined objectives reviewed by the Compensation Committee and approved by the board of directors of the Company. See “Compensation Committee” and “Report on Executive Compensation”.
41
Stock Option Incentive Compensation
The Company has in place a Stock Option Plan for its directors, officers and employees. The Stock Option Plan provides that the Board of Directors of the Company may from time to time in its discretion grant to directors, officers and employees of the Company options to purchase Common Shares. The Board of Directors determine the exercise price per Common Share and the number of Common Shares which may be allotted to each designated director, officer or employee and all other terms and conditions of the option, in accordance with the applicable policies of any relevant regulatory authority. These options are exercisable for a period not exceeding five years from the date of grant. The Board of Directors has reserved for issuance under the Stock Option Plan an aggregate of 12,000,000 of the Common Shares issued and outstanding from time to time.
The following tables set forth the options granted to and exercised by each of the Named Executives during the year ended May 31, 2002:
Option/SAR Grants During the Most Recently Completed Financial Year
| % of Total | ||||||||||||||||||||
| Options/SARs | Market Value of | |||||||||||||||||||
| Securities Under | Granted to | Securities Underlying | ||||||||||||||||||
| Options/SARs | Employees in | Exercise or | Options/SARs on the | |||||||||||||||||
| Granted | Financial | Base Price | Date of Grant | Expiration | ||||||||||||||||
| Name | (#) | Year | ($/Security) | ($/Security) | Date | |||||||||||||||
| Dr. Jim A. Wright Chief Executive Officer |
300,000 | 10.94 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06(2) | ||||||||||||
| Dr. Raafat Fahim Former President and Chief Operating Officer |
1,500,000 | 54.69 | % | $ | 0.87 | $ | 0.87 | 30-Sep-06(1) | ||||||||||||
| Dr. Aiping Young | 75,000 | 2.73 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| Senior Vice President, Research and Development | 125,000 | 4.56 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06(2) | ||||||||||||
| Mr. Geoffrey Collett | 75,000 | 2.73 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| Vice President, Corporate Development | 150,000 | 5.47 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06(2) | ||||||||||||
Mr. James T. Parsons
|
75,000 | 2.73 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06 | ||||||||||||
Vice President, Finance and Administration and CFO |
25,000 | 0.91 | % | $ | 0.95 | $ | 0.95 | 17-Sep-06(2) | ||||||||||||
| (1) | Dr. Raafat Fahim was granted options to purchase 1,500,000 common shares on October 1, 2001 at the time of hire. 1,000,000 options expired upon his resignation from his position as President and Chief Operating Officer on July 31, 2002. The remaining 500,000 options expire July 31, 2003. | |
| (2) | The officers were granted special incentive options to purchase common shares of the Company. The options vest immediately upon the attainment of specific undertakings; failing to achieve the undertakings will result in forfeiture on the specified deadline. |
Other than as described in the above footnotes, the foregoing options were granted on September 18, 2001 in respect of corporate and personal performance during the year ended May 31, 2002. The options vest on the basis of 50% on the first anniversary and 25% on the second and third anniversary of the date of granting. The exercise price of all the $0.95 options was the closing price of the Company’s common shares on the Toronto Stock Exchange on September 17, 2001.
42
Aggregated Option/SAR Exercises During the Most Recently Completed
Financial Year and Financial Year-End Option/SAR Values
| Name | Securities Acquired on Exercise (#) |
Aggregate Value Realized ($) |
Unexercised Options/SARs at May 31, 2002 (#) Exercisable/ Unexercisable |
Value
of Unexercised in-the-Money Options/SARs at May 31, 2002 ($) Exercisable/ Unexercisable |
||||
| Dr. Jim A. Wright Chief Executive Officer |
Nil | Nil | 75,000/375,000 | Nil/Nil | ||||
| Dr. Raafat Fahim Former President and Chief Operating Officer |
Nil | Nil | Nil/1,500,000 | Nil/Nil | ||||
| Dr. Aiping Young Senior Vice President, Research and Development |
Nil | Nil | 81,649/281,648 | Nil/Nil | ||||
| Mr. Geoffrey Collett Vice President, Corporate Development |
Nil | Nil | 125,000/250,000 | Nil/Nil | ||||
| Mr. James T. Parsons Vice President, Finance and Administration and CFO |
Nil | Nil | 140,000/130,000 | Nil/Nil |
Employees (the “Employees”) of the Company, other than Executive Officers, participate in a separate performance bonus plan which provides for the annual granting of options to purchase common shares under the Company’s Stock Option Plan. The Company also grants options to purchase common shares to certain employees upon commencement of their employment with the Company. During the year ended May 31, 2002, the Company granted options to Employees to purchase 229,491 common shares, being 8.4% of the total incentive stock options granted under the Stock Option Plan during the year to Employees and Executive Officers.
Pension Plan
Currently, the Company does not maintain a pension plan for its Executive Officers or its Employees. Instead of a pension plan, the Company maintains a Stock Option Plan which is available for its executive officers and employees. See “Stock Option Incentive Compensation” above. Also, the Company has a Deferred Profit Sharing Plan (“DPSP”) matching program which is available to all employees. The DPSP matching program provides 100% matching of employee contributions into the employee’s Group RRSP account up to a maximum of three percent (3%) of the employee’s gross earnings. Contributions made by the Company began in fiscal 1998 and were made to the employees’ Group Retirement Savings Plan. From February 2001, the Company’s contributions were paid into an employer-sponsored DPSP.
Directors’ and Officers’ Alternate Compensation Plan
The Company has created a directors’ and officers’ alternate compensation plan (the “Alternate Compensation Plan”). Under the Alternate Compensation Plan, the Company has the option of paying annual fees (the “Annual Fees”) to directors who are not full time employees of the Company (“Participating Directors”) by the allotment and issuance from treasury to each of the Participating Directors of such number of common shares as is equivalent to the cash value of the Annual Fees. Under the Alternate Compensation Plan, the Compensation Committee may at any time during the period between annual meetings of the
43
shareholders of the Company recommend the allotment and issuance of common shares from treasury in satisfaction of Annual Fees otherwise payable in cash to Participating Directors. The board of directors may then formally allot and issue the common shares at an issue price equal to the closing price of the common shares of the Company on the Toronto Stock Exchange on the day of, or the day immediately preceding such recommendation by the Compensation Committee or such other amount as determined by the board of directors and permitted by the stock exchange, exchanges or other market upon which the common shares are from time to time listed for trading and any other applicable regulatory authority (collectively, the “Regulatory Authorities”).
In addition, the Alternate Compensation Plan permits the Company, at its option, to satisfy the meeting attendance fees (the “Meeting Fees”) earned by the Participating Directors as a result of attendance at meetings of the board of directors held between annual meetings of the Company’s shareholders by the allotment and issuance of common shares at an issue price equal to the closing price of the common shares on the Toronto Stock Exchange on the day of, or the day immediately preceding such recommendation by the Compensation Committee or such other amount as determined by the board of directors and permitted by the Regulatory Authorities.
The Alternate Compensation Plan also permits the Company, at its option, to provide for the payment of all or part of the performance bonuses (the “Performance Bonuses”) for certain employees of the Company (the “Participating Employees”), which bonuses would otherwise be payable entirely in cash, through the issuance of common shares. Under this aspect of the Alternate Compensation Plan, the Compensation Committee may at any time recommend the allotment and issuance of common shares from treasury to one or more Participating Employees in satisfaction of Performance Bonuses established in accordance with criteria set by the board of directors (in consultation with the Compensation Committee). The issue price for the common shares will be at a price equal to the closing price of the common shares of the Company on the Toronto Stock Exchange on the day of, or the day immediately preceding such recommendation by the Compensation Committee or such other amount as determined by the board of directors and permitted by the Regulatory Authorities. The board of directors will not be obliged to allot and issue common shares to Participating Employees. However, if the board of directors elects to meet its obligations to satisfy Performance Bonuses through the allotment and issuance of common shares, it will do so by passing a resolution allotting and issuing the appropriate number of common shares only if and when the criteria for payment of the related Performance Bonuses are met.
The Alternate Compensation Plan is administered by the board of directors (in consultation with the Compensation Committee) and, subject to regulatory requirements, may be amended by the board of directors without shareholder approval, provided that the maximum number of common shares which may be issued under the Alternate Compensation Plan is not in excess of 2,500,000 common shares. common shares issued under the Alternate Compensation Plan will be subject to trading or resale restrictions under any applicable laws. The board of directors may terminate the Alternate Compensation Plan any time before or after any reservation, allotment or issuance of common shares thereunder.
Directors’ and Officers’ Deferred Share Unit Plan
The Company has created a deferred share unit plan for directors and officers (the “Deferred Share Unit Plan”). Under the Deferred Share Unit Plan, participating directors may elect to receive a portion or all of their Annual Fees from the Company in deferred share units. Under the Deferred Share Unit Plan, the Compensation Committee may at any time during the period between annual meetings of shareholders of the
44
Company, recommend the Company credit to each Participating Director who has elected under the terms of the Deferred Share Unit Plan, the number of units equal to the gross amount of the Annual Fees to be deferred divided by the fair market value of the shares. The fair market value of the shares is determined as the closing price of the common shares of the Company on the Toronto Stock Exchange on the day immediately preceding such recommendation by the Compensation Committee or such other amount as determined by the board of directors and permitted by the Regulatory Authorities.
In addition, the participating directors may elect under the Deferred Share Unit Plan to receive deferred share units in satisfaction for Meeting Fees earned by the Participating Directors as a result of attendance at meetings of the board of directors held between annual meetings of the Company’s shareholders by the credit to each Participating Director of the number of units equal to the gross amount of the Meeting Fees to be deferred divided by the fair market value of the shares, being the closing price of the common shares on the Toronto Stock Exchange on the day immediately preceding the recommendation by the Compensation Committee or such other amount as determined by the board of directors and permitted by the Regulatory Authorities.
The Deferred Share Unit Plan is administered by the board of directors (in consultation with the Compensation Committee) and, subject to regulatory requirements, may be amended by the board of directors without shareholder approval. When a Participating Director ceases to hold the position of director and is no longer otherwise employed by the Company, the Participating Director receives either (a) a lump sum cash payment equal to the number of deferred share units held multiplied by the then fair market value of the common shares of the Company on the date of termination or (b) the number of common shares that can be acquired in the open market with the amount described in (a), either case being subject to withholding for income tax. The board of directors may terminate the Deferred Share Unit Plan any time before or after any allotment or accrediting of deferred share units thereunder.
Termination of Employment, Change in Responsibilities and Employment Contracts
The Company has employment agreements (the “Agreements”) with each of the Named Executives.
The Agreements for Mr. Collett and Mr. Parsons provide for notice periods in the event of termination without cause equal to six months, plus one additional month for each year of completed service after the first year of employment. The Agreements for Dr. Wright and Dr. Young provide for a notice period of 12 months.
Dr. Fahim resigned as an employee of the Company effective July 31, 2002.
C. Board Practices
Composition of the Corporate Governance and Compensation Committee
The board of directors, upon the advice of the Compensation Committee of the board, determines executive compensation. The Compensation Committee was comprised of two directors who were not employees or officers of the Company during the period June 1, 2001 to May 31, 2002. The members of the Compensation Committee during the above period were: Peter Campbell, Barry Reiter, Robert Béchard and Alan Steigrod. In November 2001 Mr. Béchard and Mr. Steigrod replaced Mr. Campbell and Mr. Reiter as
45
members of the Compensation Committee and Mr. Béchard was appointed Chair of the Compensation Committee. The Compensation Committee met four times during the above period.
Report on Executive Compensation
The Compensation Committee’s mandate is to review, and advise the board of directors on, the recruitment, appointment, performance, compensation, benefits and termination of Executive Officers. The Compensation Committee also administers and reviews procedures and policies with respect to the Company’s Stock Option Plan, employee benefit programs, pay equity and employment equity. The philosophy of the Compensation Committee towards Executive Officer compensation is to reward performance and to provide a total compensation package that will attract and retain qualified, motivated and achievement oriented Executive Officers.
The Compensation Committee attempts to create compensation arrangements that will align the interests of the Executive Officers and the shareholders of the Company. The key components of Executive Officer compensation are base salary, potential annual cash bonuses and annual participation in the Stock Option Plan.
Base Salary — Initial Stock Options
Base salary for each Executive Officer is a function of the individual’s experience, past performance and anticipated future contribution. The Compensation Committee uses private and public compensation surveys to assist with the determination of an appropriate compensation package for each Executive Officer.
Executive Officers are granted stock options on the commencement of employment with the Company in accordance with the responsibility delegated to each Executive Officer for achieving corporate objectives and enhancing shareholder value.
Potential Annual Cash Bonuses and Annual Participation in the Stock Option Plan
Generally, potential annual cash bonuses and annual awards of options under the Stock Option Plan for each Executive Officer are conditional in part upon the achievement by the Company of predetermined scientific, clinical, regulatory, intellectual property, business and corporate development and financial objectives, and in part upon the achievement by each Executive Officer of individual performance objectives. Executive Officer individual performance objectives for each fiscal year are consistent with corporate objectives and each Executive Officer’s role in achieving them. All Corporate and Executive Officer objectives are predetermined by the board of directors after review by the Compensation Committee. Seventy-five percent (75%) of each Executive Officer’s potential annual cash bonus is conditional upon the achievement of corporate objectives with the remaining twenty-five percent (25%) being conditional upon the achievement of individual Executive Officer objectives. The Compensation Committee reserves the right to recommend to the board of directors the awarding of bonuses, payable in cash, stock or stock options, to reward extraordinary individual performance.
For each Executive Officer, during the year ended May 31, 2002, the potential annual cash bonuses ranged from 20% to 40% of base salary when all corporate and individual Executive Officer objectives
46
were achieved. For each Executive Officer, the potential annual cash bonuses range increased to 30% to 60% of base salary when the Corporate and individual Executive Officer objectives were significantly over-achieved.
Cash bonuses are determined as soon as practicable after the end of the fiscal year and are included in the Summary Compensation Table in the year in respect of which they are earned.
There is a potential for an annual allocation from the Company’s Stock Option Plan for each Executive Officer when all Corporate and Executive Officer objectives are achieved. The allocation of options is approved by the Compensation Committee of the Company and Options are priced using the closing market price of the Company’s common shares on the last trading day prior to the date of grant. Options to purchase common shares expire five years from the date of grant and vest over three years. The granting of options to purchase common shares is included in the Summary Compensation Table in the year that they are earned.
Chief Executive Officer Compensation
The performance of the Chief Executive Officer is measured in the following areas:
| • | attainment of predetermined corporate objectives; | ||
| • | attainment of predetermined individual objectives; | ||
| • | corporate governance; | ||
| • | financial condition; | ||
| • | human resource management; and | ||
| • | strategic positioning. |
Compensation of Directors
During the fiscal year ended May 31, 2002, each director who was not an officer of the Company or a representative of a shareholder was entitled to receive stock options and, at his election, shares, deferred share units and/or cash compensation for attendance at board committee meetings. Compensation is comprised of an annual fee of $5,000 ($15,000 to the chairman of the board) $1,000 per board meeting attended ($4,500 to the chairman of a board meeting) and $750 per audit, corporate governance, compensation or environmental committee meeting ($1,000 for the chairman of a committee) attended. In November 2001, stock options to purchase 270,000 common shares at a price of $1.47 per share expiring November 2006, were granted, in aggregate, to directors of the Company in this regard. In November 2000, stock options to purchase 150,000 common shares at a price of $2.12 per share expiring November 2005, were granted, in aggregate, to directors of the Company in this regard. In December 1999, a compensatory grant of stock options to purchase 330,000 common shares in aggregate, at a price of $0.82 per share expiring December 22, 2004, was made to directors of the Company. In addition, the Company reimbursed the directors for expenses incurred in attending meetings of the board of directors and committees of the board.
Mr. Barry J. Reiter resigned as a director of the Company on September 11, 2002. The law firm of Torys LLP, of which Barry J. Reiter is a partner, received legal fees in the amount of $376,000 for the year ended May 31, 2002 for acting as counsel to the Company on various matters.
47
DIRECTOR’S AND OFFICER’S LIABILITY
The Company purchases and maintains liability insurance for the benefit of directors and officers to cover any liability incurred by such person in such capacities. The policy provides for coverage in the amount of $10,000,000 with a deductible amount of $100,000. For the period June 1, 2001 to May 31, 2002, the premium cost of this insurance was $68,850.
INTEREST OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
Other than as set forth above under the heading “Executive Compensation”, during the financial year of the Company ended May 31, 2002, no director, senior officer or associate of a director or senior officer nor, to the knowledge of the directors or senior officers of the Company after having made reasonable inquiry, any person or company who beneficially owns, directly or indirectly, common shares carrying more than 10% of the voting rights attached to all common shares outstanding at the date hereof, or any associate or affiliate thereof, had any material interest, direct or indirect, in any material transaction of the Company, nor do any such persons have a material interest, direct or indirect, in any proposed transaction of the Company.
D. Employees
As at December 6, 2002, the Company had a staff of 40 full-time persons and two part-time persons, who are involved in research and drug development, and administration activities. Of the Company’s employees, eleven are medical doctors and/or Ph.D.s. The Company has a Medical and Scientific Advisory Board comprised of eight members who are each medical doctors or Ph.D.s. To encourage a focus on achieving long term performance, employees and members of the board of directors have the ability to acquire an ownership interest in the Company through the Company’s stock option plan.
E. Share Ownership
The following tables summarizes the common shares owned by the directors of the Company:
48
| Ownership or Control over | ||||||||
| common shares(1) | ||||||||
| Number of shares | % ownership | |||||||
| DONALD W. PATERSON, Toronto, Ontario President, Cavandale Corporation (corporate consulting) |
125,260 | * | ||||||
| ELLY REISMAN, Richmond Hill, Ontario President, Great Gulf Group |
1,443,208 | 1% | ||||||
| JIM A. WRIGHT, Oakville, Ontario Chief Executive Officer of the Company |
11,412,800 | 7.9% | ||||||
To the knowledge of the Company, no other directors or named executive officers beneficially own common shares in excess of 1% of the issued and outstanding capital of the Company.
Outstanding options owned by the Named Executive officers and directors of the Company
| # of | # of shares | Purchase | Expiry | |||||||||||||
| Optionee | options | entitled to purchase | Price | Date | ||||||||||||
Geoffrey Collett |
100,000 | 100,000 | $ | 2.70 | 14-Feb-05 | |||||||||||
| 50,000 | 50,000 | $ | 1.61 | 17-Dec-05 | ||||||||||||
| 75,000 | 75,000 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| 75,000 | 75,000 | $ | 0.95 | 7-Jul-07 | ||||||||||||
| 100,000 | 100,000 | $ | 0.33 | 25-Sep-07 | ||||||||||||
| 400,000 | 400,000 | |||||||||||||||
Raafat Fahim(1) |
500,000 | 500,000 | $ | 0.87 | 31-Jul-03 | |||||||||||
James T. Parsons |
120,000 | 120,000 | $ | 3.63 | 20-Feb-05 | |||||||||||
| 50,000 | 50,000 | $ | 1.61 | 17-Dec-05 | ||||||||||||
| 100,000 | 100,000 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| 75,000 | 75,000 | $ | 0.95 | 7-Jul-07 | ||||||||||||
| 75,000 | 75,000 | $ | 0.33 | 25-Sep-07 | ||||||||||||
| 420,000 | 420,000 | |||||||||||||||
Aiping Young |
50,000 | 50,000 | $ | 2.50 | 10-Oct-05 | |||||||||||
| 113,297 | 113,297 | $ | 1.61 | 17-Dec-05 | ||||||||||||
| 150,000 | 150,000 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| 75,000 | 75,000 | $ | 0.95 | 7-Jul-07 | ||||||||||||
| 75,000 | 75,000 | $ | 0.33 | 25-Sep-07 | ||||||||||||
| 463,297 | 463,297 | |||||||||||||||
Jim A. Wright |
150,000 | 150,000 | $ | 2.50 | 10-Oct-05 | |||||||||||
| 117,000 | 117,000 | $ | 0.58 | 25-Aug-07 | ||||||||||||
| 110,000 | 110,000 | $ | 0.95 | 17-Sep-06 | ||||||||||||
| 377,000 | 377,000 | |||||||||||||||
49
Outstanding options owned by the Named Executive officers and directors of the Company
| # of | # of shares | Purchase | Expiry | |||||||||||||
| Optionee | options | entitled to purchase | Price | Date | ||||||||||||
Robert Béchard |
30,000 | 30,000 | $ | 2.12 | 14-Nov-05 | |||||||||||
| 5,000 | 5,000 | $ | 2.40 | 7-Feb-06 | ||||||||||||
| 30,000 | 30,000 | $ | 1.47 | 26-Nov-06 | ||||||||||||
| 30,000 | 30,000 | $ | 0.57 | 13-Nov-07 | ||||||||||||
| 95,000 | 95,000 | |||||||||||||||
Donald W. Paterson |
7,500 | 7,500 | $ | 0.80 | 28-Jun-03 | |||||||||||
| 50,000 | 50,000 | $ | 0.40 | 25-Nov-04 | ||||||||||||
| 100,000 | 100,000 | $ | 0.82 | 22-Dec-04 | ||||||||||||
| 30,000 | 30,000 | $ | 2.12 | 14-Nov-05 | ||||||||||||
| 7,500 | 7,500 | $ | 2.40 | 7-Feb-06 | ||||||||||||
| 30,000 | 30,000 | $ | 1.47 | 26-Nov-06 | ||||||||||||
| 30,000 | 30,000 | $ | 0.57 | 13-Nov-07 | ||||||||||||
| 255,000 | 255,000 | |||||||||||||||
Elly Reisman |
30,000 | 30,000 | $ | 0.82 | 22-Dec-04 | |||||||||||
| 30,000 | 30,000 | $ | 2.12 | 14-Nov-05 | ||||||||||||
| 30,000 | 30,000 | $ | 1.47 | 26-Nov-06 | ||||||||||||
| 30,000 | 30,000 | $ | 0.57 | 13-Nov-07 | ||||||||||||
| 120,000 | 120,000 | |||||||||||||||
Alan Steigrod |
30,000 | 30,000 | $ | 1.47 | 26-Nov-06 | |||||||||||
| 30,000 | 30,000 | $ | 0.57 | 13-Nov-07 | ||||||||||||
| 60,000 | 60,000 | |||||||||||||||
Graham Strachan |
30,000 | 30,000 | $ | 1.47 | 26-Nov-06 | |||||||||||
| 125,000 | 125,000 | $ | 0.58 | 25-Aug-07 | ||||||||||||
| 90,000 | 90,000 | $ | 0.57 | 13-Nov-07 | ||||||||||||
| 245,000 | 245,000 | |||||||||||||||
Notes:
| (1) | Dr. Fahim resigned from the Company on July 31, 2002. |
50
Item 7. Major Shareholders and Related Party Transactions
A. Major shareholders
The following table provides information regarding the beneficial ownership of the Company’s common shares as of December 6, 2002 for each person or entity who is known to the Company to own beneficially more than 5% of the Company’s outstanding common shares. As used in the table, “beneficial ownership” means the sole or shared power to vote or direct the voting or to dispose or direct the disposition of any security. A person is deemed to be the beneficial owner of securities that can be acquired within 60 days from the date of this Annual Report. The amounts and percentages are based upon 144,412,000 common shares outstanding as of May 31, 2002.
| Percentage of Shares | ||||||||
| Name of Beneficial Owner | Shares Beneficially Owned | Beneficially Owned | ||||||
Jim Wright |
11,412,800 | 7.90 | % | |||||
B. Related Party Transactions
During the year ended May 31, 2002, consulting fees of $68,000 were paid to individuals (or companies controlled by those individuals) who were either officers or directors of the Company (2001 — nil and 2000 — nil).
The Company received services from a law firm in which a former director of the Company is a partner. Fees related primarily to consultations in the normal course of business for an aggregate of $376,000 for the year ended May 31, 2002 (2001 — $357,000 and 2000 — $425,000).
The amount payable to related parties as at May 31, 2002 was $46,000 (2001 — $140,000 and 2000 — $179,000).
C. Interests of Experts and Counsel
Not applicable.
Item 8. Financial Information
A. Consolidated Statements and Other Financial Information
For the Company’s consolidated financial statements, see “Item 17 Financial Statements”.
There are currently no outstanding material legal proceedings to which the Company is a party or any of the Company’s properties is subject, nor does the Company know if there are any material threatened or contemplated proceedings against it.
51
B. Significant Changes
Since May 31, 2002, there have been no significant changes in the Company’s financial condition or results of operations.
Item 9. The Offer and Listing
A. Offer and Listing Details
The common shares are registered shares. Computershare Trust Company of Canada acts as transfer agent in Canada.
The following tables show for the periods indicated the high and low closing market prices for trading taking place on both the OTC Bulletin Board (OTCBB) and the Toronto Stock Exchange (TSX).
| OTCBB | TSX | ||||||||
| Most recent five years | (In U.S. dollars) | (In Canadian dollars) | |||||||
Year ended May 31, 2002 |
|||||||||
High |
1.15 | 1.80 | |||||||
Low |
0.41 | 0.65 | |||||||
Year ended May 31, 2001 |
|||||||||
High |
2.38 | 3.43 | |||||||
Low |
0.73 | 1.05 | |||||||
Year ended May 31, 2000 |
|||||||||
High |
5.44 | 7.95 | |||||||
Low |
0.22 | 0.30 | |||||||
Year ended May 31, 1999 |
|||||||||
High |
0.67 | 1.00 | |||||||
Low |
0.16 | 0.26 | |||||||
Year ended May 31, 1998 |
|||||||||
High |
1.00 | 1.39 | |||||||
Low |
0.44 | 0.65 | |||||||
52
| OTCBB | TSX | ||||||||||||||||
| (In U.S. dollars) | (In Canadian dollars) | ||||||||||||||||
| High | Low | High | Low | ||||||||||||||
Most recent eight quarters |
|||||||||||||||||
Ended |
|||||||||||||||||
November 30, 2002 |
0.45 | 0.18 | 0.69 | 0.31 | |||||||||||||
August 31, 2002 |
0.72 | 0.37 | 1.09 | 0.58 | |||||||||||||
May 31, 2002 |
0.71 | 0.41 | 1.11 | 0.65 | |||||||||||||
February 28, 2002 |
0.84 | 0.65 | 1.37 | 1.03 | |||||||||||||
November 30, 2001 |
0.92 | 0.52 | 1.47 | 0.81 | |||||||||||||
August 31, 2001 |
1.15 | 0.70 | 1.80 | 1.07 | |||||||||||||
May 31, 2001 |
1.29 | 0.95 | 1.98 | 1.54 | |||||||||||||
February 29, 2001 |
1.66 | 0.73 | 2.59 | 1.05 | |||||||||||||
Most recent six months |
|||||||||||||||||
November, 2002 |
0.45 | 0.35 | 0.65 | 0.57 | |||||||||||||
October, 2002 |
0.31 | 0.20 | 0.69 | 0.34 | |||||||||||||
September, 2002 |
0.38 | 0.18 | 0.56 | 0.31 | |||||||||||||
August, 2002 |
0.51 | 0.37 | 0.71 | 0.58 | |||||||||||||
July, 2002 |
0.65 | 0.44 | 0.95 | 0.72 | |||||||||||||
June, 2002 |
0.72 | 0.47 | 1.09 | 0.75 | |||||||||||||
B. Plan of distribution
Not applicable to Form 20-F filed as an Annual Report.
C. Markets
The Company’s shares are listed on the OTC Bulletin Board under the symbol “LORFF” and on the Toronto Stock Exchange under the symbol “LOR”.
The National Association of Securities Dealers has submitted a proposal to the SEC to create a new marketplace to be called the BBX, which will eventually take the place of the OTC Bulletin Board. The proposed BBX will have qualitative listing standards but will have no minimum share price, income or asset requirements. This proposal is still being reviewed by the SEC and may be subject to further change and the Company will continue to analyze any impact the new proposed marketplace may have on the Company.
D. Selling Shareholders
Not applicable to Form 20-F filed as an Annual Report.
E. Dilution
Not applicable to Form 20-F filed as an Annual Report.
53
F. Expenses of the Issue
Not applicable to Form 20-F filed as an Annual Report.
Item 10. Additional Information
A. Share Capital
Not applicable to Form 20-F filed as an Annual Report.
B. Memorandum and Articles of Association
The Company’s Articles of Amalgamation and Bylaws, which were included as Exhibits 1.1 and 1.3 to the Company’s Registration Statement on Form 20-F, file number 0-19763, filed for March 4, 1992; Articles of Amendment changing the Company’s name which were included as Exhibits to the Company’s Annual Report on Form 20-F, file number 0-19763, filed for September 28, 1992, November 18, 1998 and November 30, 1999; are hereby incorporated by reference.
| 1. | Lorus Therapeutics Inc. was incorporated under the Business Corporations Act (Ontario) on September 5, 1986 under the name RML Medical Laboratories Inc. On October 28, 1991, RML Medical Laboratories Inc. amalgamated with Mint Gold Resources Ltd., resulting in the Company becoming a reporting issuer (as defined under applicable securities law) in Ontario, on such date. On August 25, 1992, the Company changed its name to IMUTEC Corporation. On November 27, 1996, the Company changed its name to Imutec Pharma Inc., and on November 19, 1998, the Company changed its name to Lorus Therapeutics Inc. | ||
| 2. | With respect to directors: |
| a. | A director’s power to vote on a proposal, arrangement or contract in which the director is materially interested is limited by the Bylaws and the governing statute. A Company’s director cannot vote in respect of any contract or transaction with the Company in which he is interested. |
| b. | The Directors may from time to time on behalf of the Company borrow money in such manner and amount, on such security, from such sources and upon such terms and conditions as they think fit. |
| c. | At each annual shareholders meeting the directors are elected to hold office until the close of the first annual meeting following his election. A director must be at least eighteen years of age. The number of annual terms is unrestricted. |
| 3. | Lorus is authorized to issue an unlimited number of common shares. The holders of common shares are entitled to one vote per share at meetings of shareholders, to receive such dividends as declared by us and to receive Lorus’s remaining property and assets |
54
| upon Lorus’ dissolution or winding up. Lorus’s common shares are not subject to any future call or assessment and there are no pre-emptive, conversion or redemption rights attached to such shares. | |||
| 4. | Changes to Lorus’s articles must be approved by the affirmative vote of shareholders holding 66-2/3 of the issued and outstanding shares present in person or by proxy at a duly called meeting of shareholders of the Company. | ||
| 5. | Pursuant to the rules of the TSX, annual general meetings of shareholders of the Company must be held within 6 months after the Company’s fiscal year end. Special meetings of shareholders may be held on appropriate notice. | ||
| 6. | There are no limitations on the rights to own the Company’s securities. | ||
| 7. | There are no provisions in the Company’s articles or bylaws that would have an effect of delaying, deferring or preventing a change in control of the company and that would operate only with respect to a merger, acquisition or corporate restructuring involving the company (or any of its subsidiaries). | ||
| 8. | There are no bylaw provisions governing the ownership threshold above which shareholder ownership must be disclosed. Securities law, however, requires the disclosure of ownership of shares of the Company above certain thresholds. |
C. Material Contracts
Not Applicable.
D. Exchange Controls
There is no law, regulation or governmental decree in Canada that restricts the export or import of capital, or affects the remittance of dividends, interest or other payments to a non-resident holder of common shares of the Company, other than withholding tax requirements. See “Taxation”.
There is no limitation imposed by Canadian law or by the charter or other constituent documents of the Company on the right of a non-resident of Canada to hold or vote common shares of the Company, other than as provided in the Investment Canada Act (Canada) (the “Investment Canada Act”). The following summarizes the principal features of the Investment Canada Act.
Under the Investment Canada Act, the acquisition of control of a Canadian business by a non-Canadian (which would include an entity organized in the United States) is subject either to a notification or review procedure. A non-Canadian would acquire control of the Company for purposes of the Investment Canada Act if it acquired a majority of the common shares of the Company or all or substantially all the assets of the Company. The acquisition of less than a majority but one-third or more of the common shares of the Company would be presumed to be an acquisition of control of the Company unless it could be established that the Company was not controlled in fact by the acquirer through the ownership of common shares.
55
The size of the Canadian business is measured against thresholds to determine if review or notification is required. Where the acquiror is a WTO investor (as defined in the Investment Canada Act; in general, an individual is a WTO investor if he or she is a national or a WTO member (a member of the World Trade Organization) and a Registrant or other entity is a WTO investor if it is a “WTO investor-controlled” entity, as determined in accordance with detailed rules set out in the Investment Canada Act), the transaction is reviewable only if, in the case of a direct acquisition of control, the Canadian business has gross assets of at least a specified amount, currently $179 million. Except in limited circumstances, an indirect acquisition of control of a Canadian business by a WTO investor is not reviewable. In circumstances where the acquiror is not a WTO investor, the threshold for a direct acquisition is $5 million and for an indirect acquisition is $50 million. In any case where an acquisition of control is reviewable, the acquiror must be able to demonstrate that the investment is likely to be of “net benefit to Canada”.
Where an investment is subject to notification, the non-Canadian is required to provide certain information concerning the investment to the Government prior to implementation of the investment or within 30 days thereafter; however, the Government retains the right to review any investment that is related to Canada’s cultural heritage or national identity or, if within a specified period, the federal cabinet considers it within the public interest on the recommendation of the Minister responsible for the Investment Canada Act to issue an order for the review of the investment.
E. Taxation
Certain Canadian Federal Income Tax Consequences to United States Shareholders
The following general discussion sets forth a summary of the material Canadian federal income tax consequences of acquiring, holding and disposing of common shares for a shareholder of the Company who is not resident in Canada for purposes of the Income Tax Act (Canada) (the “Canadian Tax Act”), and who is resident in the United States for purposes of the Canada-United States Income Tax Convention (1980) as amended (the “Convention”), and who, for purposes of the Canadian Tax Act, holds its common shares as capital property, deals at arm’s length with the Company, and does not use or hold and is not deemed to use or hold such common shares in connection with a business carried on in Canada.
The following discussion is based on the current provisions of the Canadian Tax Act and the regulations thereunder and on the Company’s understanding of the current administrative practices of Canada Customs and Revenue Agency and takes into account all specific proposals to amend the Canadian Tax Act or regulations made by the Minister of Finance of Canada before the date hereof. The discussion is general only and is not a substitute for independent advice from a holder’s own tax adviser.
The provisions of the Tax Act are subject to income tax treaties to which Canada is a party, including the Convention.
Dividends on common shares
Under the Canadian Tax Act, a non-resident of Canada is generally subject to Canadian withholding tax at the rate of 25% on dividends paid or credited to the non-resident by a Registrant resident in Canada. The Convention limits the rate to 15% if the shareholder is resident in the United States and the dividends are beneficially owned by him and to 5% if the shareholder is also a Registrant that owns at least 10% of the voting stock of the payer Registrant.
56
Disposition of common shares
A non-resident of Canada will not be subject to tax in Canada on any capital gain realized on a disposition of common shares, provided that the common shares do not constitute “taxable Canadian property” of the non-resident. The common shares will not generally constitute “taxable Canadian property” of a non-resident unless, at any time within the five-year period immediately preceding the disposition, the non-resident owned, had an interest in or had the right to acquire, either alone or together with persons with whom the non-resident does not deal at arm’s length, 25% of more of the issued shares of any series or class of the capital stock of the Company. If common shares are “taxable Canadian property” to a shareholder that is resident in the United States, such shareholder may be exempted from Canadian capital gains tax under the Convention.
Certain United States Federal Income Tax Considerations for United States Shareholders
The following general discussion sets forth a summary of the material U.S. federal income tax considerations that are applicable to the following persons who hold common shares as capital assets (“U.S. Shareholders”): (i) citizens or residents (as specially defined for U.S. federal income tax purposes) of the United States, (ii) companies created or organized in the United States or under the laws of the United States or of any state or the District of Columbia, (iii) estates, the income of which is subject to U.S. federal income taxation regardless of its source, and (iv) a trust whose administration is subject to the primary supervision of a U.S. court and which has one or more U.S. fiduciaries who have the authority to control all substantial decisions of the trust.
This discussion is based on current provisions of the Internal Revenue Code of 1986, as amended (the “Code”), current and proposed Treasury regulations promulgated thereunder (“Treasury Regulations”), and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly on a retroactive basis. This discussion does not address all of the aspects of U.S. federal income taxation that may be relevant to any particular shareholder’s circumstances. This discussion does not address the tax treatment of the following types of shareholders, who may be subject to special rules not discussed below: shareholders who own (directly, indirectly or through attribution) 10% or more of the Company’s outstanding voting stock, who hold common shares as a hedge or who are broker-dealers, insurance companies, tax exempt organizations, financial institutions, or taxpayers whose functional currency is not the U.S. dollar. Additionally, this summary does not address the possible application of U.S. federal gift or estate taxes, or any aspect of state, local or non-U.S. tax laws. Each U.S. Shareholder is advised to consult such person’s own tax advisor with respect to the specific U.S. federal income and other tax consequences to such person of purchasing, holding or disposing of the common shares.
Passive Foreign Investment Companies
For any taxable year of the Company, if 75% or more of the Company’s gross income (including the Company’s share of the gross income for any company in which the Company is considered to own 25% or more of the shares by value) is “passive income” (as defined in the relevant provision of the Code) or if at least 50% of the Company’s assets (including the Company’s share of the value of the assets of any Corporation in which the Company is considered to own 25% or more of the shares by value), by average fair market value, are assets that produce or are held for the production of passive income, the Company will be a Passive Foreign Investment (“PFIC”). The Company currently is a PFIC and expects to be a PFIC for the foreseeable future.
57
A U.S. Shareholder of a PFIC is subject to special U.S. federal income tax rules contained in Sections 1291 to 1297 of the Code. As described below, these provisions set forth three alternative tax regimes applicable at the election of each U.S. Shareholder, depending upon whether the U.S. Shareholder elects to treat the Company as a “qualified electing fund” (a “QEF Election”) to market (a “Mark to Market Election) or elects to mark the PAC stock.
U.S. SHAREHOLDERS ARE URGED TO CONSIDER MAKING A QEF ELECTION TO AVOID CERTAIN POTENTIALLY SIGNIFICANT ADVERSE U.S. TAX CONSEQUENCES.
1. The QEF Election Alternative
Each U.S. Shareholder is urged to consider making a QEF Election because of the potential benefits of such election that are discussed below and because the Company anticipates that it will not have any earnings and profits (as computed for U.S. federal income tax purposes) for the current taxable year and little, if any, earnings and profits for any future taxable year in which the Company is a PFIC. There can be no assurance, however, that this will be the case. Accordingly, the timely making of the QEF Election, as discussed below, generally should avoid any significant adverse U.S. federal income tax consequences resulting from any classification of the Company as a PFIC, although this may depend on a particular U.S. Shareholder’s circumstances.
A U.S. Shareholder who elects in a timely manner to treat the Company as a QEF (an “Electing U.S. Shareholder”) will be subject to federal income tax on a current basis for any taxable year in which the Company is a PFIC (or is treated as a PFIC with respect to the U.S. Shareholder) on such Electing U.S. Shareholder’s pro-rata share of the Company’s (i) “net capital gain” (the excess of net long-term capital gain over net short-term capital loss, but not in excess of the Company’s current earnings and profits), which will be taxed as long-term capital gain to the Electing U.S. Shareholder and (ii) “ordinary earnings” (the excess of earnings and profits over net capital gain), which will be taxed as ordinary income to the Electing U.S. Shareholder, in each case, for the shareholder’s taxable year in which (or with which) the Company’s taxable year ends, regardless of whether such amounts actually are distributed. Adjustments are provided generally to prevent double taxation at the time of later distributions on or dispositions of common shares.
The QEF Election also allows an Electing U.S. Shareholder (i) generally to treat any gain realized on the disposition of common shares (or deemed to be realized on the pledge of such shareholder’s common shares) as capital gain; and (ii) generally to avoid interest charges resulting from PFIC status altogether.
The Company undertakes to provide its U.S. Shareholders with timely and accurate information as to its status as a PFIC and to comply with all record keeping, reporting and other requirements so that each U.S. Shareholder may elect to treat the Company as a QEF. Such an election must be made by attaching the following documents to the timely filed U.S. federal income tax return for the first taxable year of the U.S. Shareholder in which or with which falls the end of a taxable year of the Company during which the Company was a PFIC and the U.S. Shareholder held (or was considered to have held) common shares: (i) a “Shareholder Section 1295 Election Statement” executed by the U.S. Shareholder, (ii) a “PFIC Annual Information Statement” received by the U.S. Shareholder from the Company, and (iii) a Form 8621. In addition, the Electing U.S. Shareholder must file a copy of the Shareholder Section 1295 Election Statement with the Internal Revenue Service Center, P.O. Box 21086, Philadelphia, PA 19114.
58
The following three paragraphs apply to Electing U.S. Shareholders:
Dividends Paid on common shares. Any dividends paid to an Electing U.S. Shareholder on common shares (including any Canadian taxes withheld) will be treated as ordinary dividend income for U.S. federal income tax purposes to the extent of the Company’s current and accumulated earnings and profits (as computed for U.S. federal income tax purposes) unless paid out of earnings and profits that previously were taxed to the Electing U.S. Shareholder under the QEF rules. Such dividends generally will not qualify for the dividends-received deduction otherwise available to US corporations. Amounts in excess of such earnings and profits will be applied against the Electing U.S. Shareholder’s tax basis in the common shares and, to the extent in excess of such tax basis, will be treated as gain from a sale or exchange of such common shares.
Credit for Canadian Taxes Withheld. Subject to the limitations in the Code the Canadian tax withheld or paid with respect to dividends on the common shares generally may be taken as a foreign tax credit against U.S. federal income taxes by an Electing U.S. Shareholder who chooses to claim such a credit for the taxable year. Electing U.S. Shareholders who do not choose to claim foreign tax credits for a taxable year may claim a U.S. tax deduction for such Canadian tax for that taxable year.
Disposition of common shares. An Electing U.S. Shareholder will recognize capital gain or loss for U.S. federal income tax purposes upon the sale or other disposition of common shares in an amount equal to the difference between the net amount realized on the disposition and the U.S. Shareholder’s adjusted tax basis in the common shares. In the case of non-corporate U.S. Shareholders, such gain or loss will be (i) short-term gain or loss for common shares held for one year or less, and (ii) long-term gain or loss for common shares held for more than one year. Currently, the maximum rate of U.S. federal income tax on individuals’ long-term capital gains is 20%. Capital losses of individuals may offset any capital gains plus $3,000 ($1,500 for married persons filing separately) of ordinary income. For taxable years beginning after December 31, 2000, some individuals may be entitled to lower capital gain rates with respect to common shares held for more than five years.
2. The Non-QEF Election Alternative
If a U.S. Shareholder does not make a timely QEF Election for the first taxable year of the Company during which he holds (or is considered to hold) the common shares in question and the Company is a PFIC (a “Non-Electing U.S. Shareholder”), or makes a Mark to Market Election as discussed below, then special rules under Section 1291 of the Code will apply to (i) gains realized on the disposition (or deemed to be realized by reason of a pledge) of common shares, and (ii) certain distributions by the Company (collectively, “excess distributions”). The Company has never made any distributions with respect to the common shares and it does not anticipate making any such distributions in the foreseeable future.
A Non-Electing U.S. Shareholder generally would be required to pro-rate all gains realized on the disposition of common shares, and all excess distributions, over such shareholder’s entire holding period for the common shares. All portions of excess distributions allocated to prior years of the U.S. Shareholder (provided that such periods are not prior to the first day of the first taxable year of the Company during such U.S. Shareholder’s holding period and beginning after December 31, 1986 for which it was a PFIC) would be taxed at the highest tax rates applicable to ordinary income for each such prior year. The Non-Electing U.S. Shareholder also would be liable for interest with respect to the tax liability for each such prior year, calculated as if such tax had been due with respect to each such prior year. All portions of excess distributions allocated to periods prior to the first day of the first taxable year of the Company during the U.S. Shareholder’s holding period for which it was a PFIC, and all excess distributions allocated to the current year, will be treated as
59
ordinary income in the year of the distribution and no interest charge will be incurred with respect to such amounts.
If the Company is a PFIC for any taxable year during which a Non-Electing U.S. Shareholder holds (or is considered to hold) common shares, then the Company will continue to be treated as a PFIC with respect to such common shares, even if the Company does not qualify as a PFIC in a later year. A Non-Electing U.S. Shareholder may terminate this deemed PFIC status by electing to recognize gain (which will be taxed under the rules discussed above for Non-Electing U.S. Shareholders) as if such common shares had been sold on the last day of the last taxable year for which it was a PFIC. Certain other elections are also available to Non-Electing U.S. Shareholders.
3. Mark to Market Election Alternative
Effective for taxable years ending after 1997, a U.S. Shareholder of certain publicly traded PFIC stock, including the Company, may elect to mark such stock to market annually, recognizing as ordinary income or loss each year an amount equal to the difference between the holder’s adjusted tax basis in PFIC stock and its fair market value. Losses are allowed only to the extent of net mark-to-market gain previously included by the U.S. Shareholder pursuant to elections for prior taxable years. If a mark-to-market election is made, any further gain or loss realized on a disposition of common shares will be treated as ordinary income or loss. The source of any income or losses recognized pursuant to a mark-to-market election is determined as if the amount were gain or loss from the sale of PFIC stock. The tax basis of PFIC stock for which an election is made will be increased by the income recognized pursuant to the election and decreased by the deductions allowed under the election. If the mark-to-market election is made, then the rules for Non-Electing U.S. Shareholders described in the preceding paragraphs will not apply for the periods covered by the election. The mark-to-market election applies to the taxable year for which made and to all subsequent taxable years, unless the PFIC stock ceases to be publicly traded or the Treasury Secretary consents to the revocation of the election.
Future Developments
The foregoing discussion is based on current provisions of the Code, existing and proposed regulations thereunder, and current administrative rulings and court decisions, all of which are subject to change. Any such changes could affect the validity of this discussion. In addition, the implementation of certain aspects of the PFIC rules requires the issuance of regulations which in many instances have not yet been promulgated and which may have retroactive effect.
ALL INVESTORS ARE ADVISED TO CONSULT THEIR OWN TAX ADVISORS WITH RESPECT TO THE SPECIFIC TAX CONSEQUENCES OF PURCHASING OR HOLDING COMMON SHARES.
F. Dividends and Paying Agents
Not applicable to Form 20-F filed as an Annual Report.
G. Statement by Experts
Not applicable to Form 20-F filed as an Annual Report.
60
H. Documents on Display
The documents concerning the Company which are referred to in this Annual Report are either annexed hereto as exhibits, or may be inspected at the principal executive offices of the Company at 2 Meridian Road, Toronto, Ontario, Canada, M9W 4Z7.
I. Subsidiary Information
Lorus’ subsidiaries are GeneSense, a company incorporated under the laws of Canada of which the Company owns 100% of the issued and outstanding share capital and NuChem, a company incorporated under the laws of Ontario of which Lorus owns 80% of the issued and outstanding common share capital.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
Impact of interest rate exposure
As of May 31, 2002, the Company had $37,822,000 in cash, cash equivalent and short-term investments, of which $36,657,000 were short-term investments. A significant portion of the short-term investments are debt securities. Interest income is subject to fluctuations due to changes in interest rates. Investments are held to maturity and have staggered maturities to minimize interest rate risk. The Company purchases some services and manufactured drugs in U.S. currency, and conducts clinical trials in the United States. U.S. dollar expenditures are expected to increase in 2003 with additional clinical trials beginning in the United States. the Company does not currently engage in hedging its U.S. currency requirements to reduce exchange rate risk. Exchange gain or loss realized in the past years were minimal.
Financial instruments potentially expose the Company to a concentration of credit risk. The Company’s financial instruments consist principally of cash equivalents and short-term investments. The Company mitigates the risk by investing in high grade fixed income securities.
Item 12. Description of Securities Other than Equity Securities
Not applicable to Form 20-F filed as an Annual Report.
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds
Not applicable.
Item 15. [Reserved]
61
Item 16. [Reserved]
PART III
Item 17. Financial Statements
The financial statements of the Company have been prepared on the basis of Canadian GAAP and in all material respects comply with U.S. GAAP.
The auditors’ report, financial statements and notes thereto, schedules thereto as required under Item 17 are found immediately below. The Audit Report of KPMG LLP, Chartered Accountants, is included herein immediately preceding the respective financial statements, notes, schedules, etc.
62
TABLE OF CONTENTS
| Sections | Page # | |||
Management’s responsibility for Financial statements |
64 | |||
Auditor’s report |
65 | |||
Balance Sheet |
66 | |||
Statement of loss and deficit |
67 | |||
Statement of cash flow |
68 | |||
Notes to the financial statements |
69-80 | |||
63
MANAGEMENT’S RESPONSIBILITY FOR FINANCIAL STATEMENTS
The accompanying consolidated financial statements and all information in this annual report have been prepared by management and have been approved by the Board of Directors.
The financial statements have been prepared in accordance with Canadian generally accepted accounting principles and include amounts that are based on the best estimates and judgements of management. Financial information presented elsewhere in the annual report is consistent with that in the financial statements.
The integrity and objectivity of these financial statements are the responsibility of management. In support of this responsibility, management maintains a system of internal controls to provide reasonable assurance as to the reliability of financial information and the safeguarding of assets.
The Audit Committee reviews the consolidated financial statements, adequacy of internal controls, audit process and financial reporting with management and with the external auditors. The Audit Committee, which consists of three directors not involved in the daily operations of the Company, reports to the Board of Directors prior to the approval of the audited consolidated financial statements for publication.
The external auditors have free and full access to the Audit Committee with respect to their findings concerning the fairness of financial reporting and the adequacy of internal controls. These financial statements have been audited by the shareholders’ independent auditors, KPMG LLP.
| Jim A. Wright | James T. Parsons | |
| Chief Executive Officer |
VP Finance and Administration and Chief Financial Officer |
|
| June 28, 2002 |
64

| KPMG LLP | ||||
| Chartered Accountants | Telephone | (416)228-7000 | ||
| Yonge Corporate Centre | Telefax | (416)228-7123 | ||
| 4100 Yonge Street Suite 200 | www.kpmg.ca | |||
| Toronto ON M2P 2H3 | ||||
| Canada |
AUDITORS’ REPORT TO THE SHAREHOLDERS
We have audited the consolidated balance sheets of Lorus Therapeutics Inc. as at May 31, 2002 and 2001 and the consolidated statements of loss and deficit and cash flows for each of the years in the three-year period ended May 31, 2002 and the related consolidated statements of loss and deficit and cash flows for the period from inception on September 5, 1986 to May 31, 2002. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform an audit to obtain reasonable assurance whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, these consolidated financial statements present fairly, in all material respects, the financial position of the Company as at May 31, 2002 and 2001 and the results of its operations and its cash flows for each of the years in the three-year period ended May 31, 2002 and for the period from inception on September 5, 1986 to May 31, 2002 in accordance with Canadian generally accepted accounting principles.
We did not audit the consolidated financial statements of Lorus Therapeutics Inc. for the period from inception on September 5, 1986 to May 31, 1994. Those consolidated financial statements were audited by other auditors who issued a report without reservation on July 8, 1994.
Chartered Accountants
Toronto, Canada
June 28, 2002
65
Lorus Therapeutics Inc.
Consolidated Balance Sheets
As at May 31
| (Amounts in 000's) (Canadian Dollars) | 2002 | 2001 | |||||||||
ASSETS |
|||||||||||
Current assets |
|||||||||||
Cash and cash equivalents |
$ | 1,165 | $ | 2,783 | |||||||
Short-term investments |
36,657 | 46,035 | |||||||||
Prepaid expenses and amounts receivable |
1,195 | 1,504 | |||||||||
Total current assets |
39,017 | 50,322 | |||||||||
Fixed assets (note 4) |
533 | 262 | |||||||||
Goodwill (note 3 (a)) |
606 | 2,060 | |||||||||
Acquired research and development (notes 5 and 8) |
7,416 | 9,163 | |||||||||
| $ | 47,572 | $ | 61,807 | ||||||||
LIABILITIES AND SHAREHOLDERS’ EQUITY |
|||||||||||
Current liabilities |
|||||||||||
Accounts payable |
$ | 442 | $ | 3,128 | |||||||
Accrued liabilities |
2,990 | 2,737 | |||||||||
Total current liabilities |
3,432 | 5,865 | |||||||||
Shareholders’ equity |
|||||||||||
Share capital (note 6) |
|||||||||||
Common shares |
|||||||||||
Authorized: unlimited number of shares; |
|||||||||||
Issued and outstanding (000’s): |
|||||||||||
May 31,
2002 — 144,412 |
|||||||||||
May 31, 2001 — 142,411 |
119,168 | 117,150 | |||||||||
Warrants |
— | 729 | |||||||||
Deferred stock-based compensation (note 6 (h)) |
(159 | ) | (555 | ) | |||||||
Deficit accumulated during development stage |
(74,869 | ) | (61,382 | ) | |||||||
Total shareholders’ equity |
44,140 | 55,942 | |||||||||
| $ | 47,572 | $ | 61,807 | ||||||||
Commitments (Notes 3(b) and 10)
Canada and United States accounting policy differences (note 13)
See accompanying notes to consolidated financial statements
66
Lorus Therapeutics Inc.
Consolidated Statements of Loss and Deficit
| Period from | ||||||||||||||||
| Years Ended May 31 | inception | |||||||||||||||
| (Amounts in 000's except for per common share data) | Sept. 5, 1986 to | |||||||||||||||
| (Canadian Dollars) | 2002 | 2001 | 2000 | May 31, 2002 | ||||||||||||
EXPENSES |
||||||||||||||||
Research and development (note 8) |
$ | 8,659 | $ | 9,797 | $ | 4,244 | $ | 46,509 | ||||||||
General and administrative |
4,867 | 6,414 | 3,652 | 28,588 | ||||||||||||
Depreciation and amortization |
1,956 | 1,903 | 1,245 | 7,401 | ||||||||||||
Interest income |
(1,995 | ) | (2,901 | ) | (542 | ) | (7,629 | ) | ||||||||
Loss for the period |
13,487 | 15,213 | 8,599 | 74,869 | ||||||||||||
Deficit, beginning of period |
61,382 | 46,169 | 37,570 | — | ||||||||||||
Deficit, end of period |
$ | 74,869 | $ | 61,382 | $ | 46,169 | $ | 74,869 | ||||||||
Basic and diluted loss
per common share (note 2) |
$ | 0.09 | $ | 0.11 | $ | 0.10 | ||||||||||
Weighted average number of
common shares outstanding
used in the calculation of basic
and diluted loss per share |
143,480 | 140,776 | 86,121 | |||||||||||||
See accompanying notes to consolidated financial statements
67
Lorus Therapeutics Inc.
Consolidated Statements of Cash Flows
| Period from | |||||||||||||||||
| Years Ended May 31 | inception | ||||||||||||||||
| (Amounts in 000's) | Sept. 5, 1986 to | ||||||||||||||||
| (Canadian Dollars) | 2002 | 2001 | 2000 | May 31, 2002 | |||||||||||||
OPERATING ACTIVITIES |
|||||||||||||||||
Loss for the period |
$ | (13,487 | ) | $ | (15,213 | ) | $ | (8,599 | ) | $ | (74,869 | ) | |||||
Add items not requiring a
current outlay of cash: |
|||||||||||||||||
Depreciation and amortization |
3,703 | 3,703 | 2,662 | 12,590 | |||||||||||||
Other |
— | — | — | 500 | |||||||||||||
Net change in non-cash working capital
balances related to operations (note 9) |
(2,124 | ) | 1,848 | 575 | 1,330 | ||||||||||||
Cash used in operating activities |
(11,908 | ) | (9,662 | ) | (5,362 | ) | (60,449 | ) | |||||||||
INVESTING ACTIVITIES |
|||||||||||||||||
Sale (purchase) of short-term investments, net |
9,378 | (40,376 | ) | (5,659 | ) | (36,657 | ) | ||||||||||
Acquisition, net of cash received (note 3(a)) |
— | — | (539 | ) | (539 | ) | |||||||||||
Acquired research and development |
— | — | — | (715 | ) | ||||||||||||
Additions to fixed assets |
(477 | ) | (172 | ) | (19 | ) | (3,732 | ) | |||||||||
Cash proceeds on sale of fixed assets |
— | — | 116 | 348 | |||||||||||||
Cash provided by (used in)
investing activities |
8,901 | (40,548 | ) | (6,101 | ) | (41,295 | ) | ||||||||||
FINANCING ACTIVITIES |
|||||||||||||||||
Issuance of warrants |
— | — | 9,512 | 31,877 | |||||||||||||
Issuance of common shares |
1,389 | 2,065 | 51,592 | 71,032 | |||||||||||||
Cash provided by financing activities |
1,389 | 2,065 | 61,104 | 102,909 | |||||||||||||
Increase (decrease) in cash and cash
equivalents during the period |
(1,618 | ) | (48,145 | ) | 49,641 | 1,165 | |||||||||||
Cash and cash equivalents,
beginning of period |
2,783 | 50,928 | 1,287 | — | |||||||||||||
Cash and cash equivalents,
end of period |
$ | 1,165 | $ | 2,783 | $ | 50,928 | $ | 1,165 | |||||||||
See accompanying notes to consolidated financial statements
68
For the years ended May 31, 2002, 2001 and 2000
1. Description of Business
Lorus Therapeutics Inc. (“Lorus” or “the Company”) is a biopharmaceutical company specializing in the research, development and commercialization of pharmaceutical products and technologies for the management of cancer. With products in all stages of evaluation, from pre-clinical through Phase III trials, and a product approved in Mexico for malignant melanoma, Lorus develops therapeutics that seek to manage cancer with efficacious non-toxic compounds that improve patients’ quality of life.
2. Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the accounts of Lorus, its 80% owned subsidiary NuChem Pharmaceuticals Inc. (“NuChem”), and its wholly-owned subsidiary GeneSense Technologies Inc. (“GeneSense”). The results of operations for acquisitions are included in these consolidated financial statements from the date of acquisition. All significant intercompany balances and transactions have been eliminated on consolidation. The consolidated financial statements have been prepared by management in accordance with accounting principles generally accepted in Canada and comply in all material respects with accounting principles generally accepted in the United States, except as disclosed below under “Recent Accounting Pronouncements” and in note 13 “Canada and United States Accounting Policy Differences”.
Cash Equivalents and Short-Term Investments
Lorus invests in high quality government and corporate issuers with low credit risk. Cash equivalents consist of highly liquid investments with a maturity of three months or less at the time of purchase. Short-term investments, which consist of fixed income securities with a maturity of three months or more, are recorded at their accreted value as they are held to maturity instruments.
Fixed Assets
Fixed assets are recorded at cost. The Company provides depreciation and amortization at rates which are expected to charge operations with the cost of the assets over their estimated useful lives as follows:
| Furniture and equipment Leasehold improvements | straight-line over three to five years straight-line over the lease term |
69
The Company regularly reviews the carrying value of its fixed assets by comparing the carrying amount of the assets to the expected future cash flows to be generated by the assets. If the carrying value exceeds the amount recoverable, a write-down is charged to the statement of operations.
Research and Development
Research costs are charged to expense as incurred. Development costs, including the cost of drugs for use in clinical trials, are expensed as incurred unless they meet the criteria under generally accepted accounting principles for deferral and amortization. No development costs have been deferred to date.
The Company capitalized the cost of acquired research and development on the acquisitions of GeneSense and the NuChem compounds and is amortizing these costs on a straight-line basis over seven years. Management reviews the carrying value of acquired research and development and accounts for any permanent impairment in value as a charge to operations in the year incurred.
The carrying value of acquired research and development does not necessarily reflect its present or future value. The amount recoverable is dependent upon the continued advancement of the drugs through research, clinical trials and ultimately to commercialization. It is not possible to predict the outcome of future research and development programs.
The Company has not earned revenues from its drug candidates and is therefore considered to be in the development stage.
Goodwill
Goodwill represents the excess of the cost of the GeneSense acquisition over the fair value of the net assets acquired and is being amortized on a straight line basis over three years. Management reviews the carrying value of goodwill and accounts for any permanent impairment in value as a charge to operations in the year incurred. Commencing June 1, 2002, the Company will adopt the new accounting standard related to goodwill as described below under “Recent Accounting Pronouncements”.
Stock-Based Compensation
Stock options granted to employees are accounted for using the intrinsic value method. Under the intrinsic value method, compensation cost is recorded if, on the measurement date of the grant, the fair value of an underlying common share exceeds the exercise price per share. For options with contingent vesting criteria, the option is treated as a variable award and is revalued, using the intrinsic value method of accounting, at the end of each reporting period until the final measurement date. Compensation cost is amortized over the vesting period of the option.
The Company has a deferred share unit plan that provides directors the alternative to receive payment for their current services in the form of share units rather than common shares or cash. Share units entitle the holder to receive, in the future, either an equivalent number of common shares or the cash equivalent of the shares at the date the units are exercised. As the award entitles the holder to settle the award through the receipt of cash, the value of the share units are recorded as a liability and the share units are revalued each reporting date with any increase or decrease in value being recorded in the consolidated statement of loss.
70
Stock options granted to consultants and other non-employees are accounted for using the fair value method. Under this method, options granted are recognized at their fair value as services are performed and/or options are earned.
Income Taxes
Income taxes are reported using the asset and liability method, where future tax assets and liabilities are recorded for the future tax consequences attributable to differences between the financial statement carrying amounts of assets and liabilities and their respective tax bases, and operating loss and research and development expenditure carryforwards. Future tax assets and liabilities are measured using enacted or substantially enacted tax rates expected to apply when the asset is realized or the liability is settled. The effect on future tax assets and liabilities of a change in tax rates is recognized in income in the period that substantive enactment or enactment occurs. A valuation allowance is recorded for the portion of the future tax assets where the realization of any value is uncertain.
Earnings Per Share
On June 1, 2001, the Company adopted, on a retroactive basis, the new accounting recommendations of the Canadian Institute of Chartered Accountants with respect to calculating loss per share. Basic net loss per common share is based on the weighted average number of shares outstanding during each period.
Under the new recommendations, the treasury stock method is used in the calculation of dilutive loss per common share instead of the previously applied imputed earnings approach for determining the effect of all dilutive elements. The adoption of the new method had no effect on the diluted loss per share because there are no dilutive elements under either standard. Stock options and warrants are not included in the computation of the weighted average number of shares outstanding for dilutive net loss per common share when the effect would be anti-dilutive.
Segmented Information
The Company is organized and operates as one operating segment, the research and development of cancer therapies.
Use of Estimates
The preparation of financial statements requires management to make estimates and assumptions that affect the amounts presented in the financial statements and the accompanying notes. Actual results could differ from these estimates.
Foreign Currency Translation
Foreign currency transactions are translated into Canadian dollars at rates prevailing on the transaction dates. Monetary assets and liabilities are translated into Canadian dollars at the rates on the balance sheet dates. Gains or losses resulting from these transactions are accounted for in the loss for the period and are not significant.
71
Recent Accounting Pronouncements
In August 2001, the Canadian Accounting Standards board (“AcSB”) issued Handbook Section 1581, “Business Combinations”, and Handbook Section 3062, “Goodwill and Other Intangible Assets”. Section 1581 requires that all business combinations be accounted for by the purchase method and it sets out criteria in determining the valuation and allocation of the purchase price in a business combination to tangible assets, intangible assets and goodwill. Section 3062 requires that goodwill no longer be amortized to earnings, but instead be periodically reviewed for impairment. Section 3062 also requires that intangible assets be assessed to determine if they have an estimated useful life or whether they have an indefinite life. Intangible assets that have an estimated useful life will continue to be amortized systematically over the useful life. Intangible assets with indefinite useful lives are not to be amortized but are instead to be tested for impairment annually. Upon adoption of the new Section 3062 in fiscal 2003, the Company must perform transitional impairment tests on goodwill and intangible assets with indefinite lives. Any impairment losses are to be measured as of the date of adoption. Impairment losses assessed on transition, if any, will be recorded as an adjustment to retained earnings. The impact of adopting Section 1581 and 3062 has not yet been determined.
In July 2001, the U.S. Financial Accounting Standards Board (“AcSB”) issued Statement of Financial Accounting Standard (“SFAS”) No. 141, “Business Combinations” and SFAS 142 “Goodwill and Other Intangible Assets” which are consistent with Sections 1581 and 3062, respectively, except for certain remaining generally accepted accounting principles (“GAAP”) differences, including the accounting for purchased in-process research and development and the recording of any impairment charge determined on transition as a period cost which are required under U.S. GAAP.
In December 2001, the AcSB issued Handbook Section 3870 “Stock-Based Compensation and Other Stock-Based Payments”. Section 3870 establishes standards for the recognition, measurement, and disclosure of stock-based compensation and other stock-based payments made in exchange for goods and services provided by employees and non-employees. It applies to transactions in which common shares, stock options, or other equity instruments are granted or liabilities incurred based on the price of common stock or other equity instruments.
The Company will adopt Section 3870 for its fiscal year beginning June 1, 2002. The Company does not believe that the adoption of this standard will have a material impact on the Company’s financial condition or results of operations as the Company’s current accounting policies, as disclosed above, comply with the new standard.
3. Acquisitions
| (a) In October 1999, the Company completed the acquisition of all of the issued and outstanding shares of GeneSense Technologies Inc. a molecular genetic drug development company specializing in oligonucleotide therapies for the treatment of cancer and infectious diseases. |
72
The acquisition was accounted for using the purchase method. The total cost of the acquisition of $14,775,000 was allocated to the fair value of the net assets acquired as follows:
| Amounts in 000's | ||||
Current assets |
$ | 822 | ||
Fixed assets |
83 | |||
Acquired research and development |
11,000 | |||
Goodwill |
4,363 | |||
Current liabilities |
(1,493 | ) | ||
| $ | 14,775 | |||
The purchase price was satisfied by the issuance of 36,050,000 Lorus common shares. In addition, the Company issued 7,210,000 common share purchase warrants and 903,825 employee stock options in exchange for 1,400,000 common share purchase warrants and 175,500 employee stock options of GeneSense which were outstanding immediately prior to the acquisition. The purchase warrants entitled the holder to acquire one common share of Lorus for $0.6932 per share. The employee stock options have an exercise price of $0.40 per common share and maintain their original vesting terms. The total purchase price includes $775,000 in cash paid for costs related to the acquisition. All common share purchase warrants issued in connection with the acquisition were exercised in the 2001 fiscal year for proceeds of $4,998,000.
| (b) In December 1997, NuChem acquired certain patent rights and a sub-license to develop and commercialize the anti-cancer application of certain compounds in exchange for a 20% share interest in NuChem, the payment of US$350,000 in shares of Lorus, and up to US$3,500,000 in cash. In 1999, the Company issued 583,188 common shares from treasury in settlement of the US$350,000 and made cash payments of US$500,000 (Cdn. $715,000). The remaining balance of up to US$3,000,000 remains payable upon the achievement of certain milestones based on the commencement and completion of clinical trials. The payments made to date of $1,228,000 have been classified as acquired research and development. Lorus funds all research and development expenses of NuChem. |
4. Fixed Assets
| As at May 31 (amounts in 000's) | 2002 | 2001 | ||||||
Furniture and equipment |
$ | 1,171 | $ | 765 | ||||
Leasehold improvements |
139 | 68 | ||||||
| 1,310 | 833 | |||||||
Accumulated depreciation and
amortization |
(777 | ) | (571 | ) | ||||
| $ | 533 | $ | 262 | |||||
5. Acquired Research and Development
| As at May 31 (amounts in 000's) | 2002 | 2001 | ||||||
Cost |
$ | 12,228 | $ | 12,228 | ||||
Accumulated amortization |
(4,812 | ) | (3,065 | ) | ||||
| $ | 7,416 | $ | 9,163 | |||||
73
6. Share Capital
| (a) Continuity of common shares and warrants |
| Common Shares | Warrants | |||||||||||||||||||
| Amounts in 000's | Note 6 | Number | Amount | Number | Amount | |||||||||||||||
Balance at May 31, 1999 |
42,747 | $ | 38,955 | 4,093 | $ | 535 | ||||||||||||||
Exercise of purchase warrants |
(b) | 893 | 1,821 | (893 | ) | (321 | ) | |||||||||||||
Exercise of purchase warrants |
(c) | 3,200 | 1,333 | (3,200 | ) | (213 | ) | |||||||||||||
Issuance of special and purchase warrants |
(d) | — | — | 33,128 | 8,853 | |||||||||||||||
Exercise of special warrants |
(d) | 30,303 | 8,438 | (30,303 | ) | (8,438 | ) | |||||||||||||
Exercise of purchase warrants |
(d) | 2,181 | 1,215 | (2,181 | ) | (321 | ) | |||||||||||||
Issuance in public offering |
(e) | 15,333 | 41,952 | 766 | 659 | |||||||||||||||
Issued on acquisition of GeneSense
(note 3 (a)) |
36,050 | 14,000 | 7,210 | — | ||||||||||||||||
Exercise of purchase warrants (note 3 (a)) |
7,210 | 4,998 | (7,210 | ) | — | |||||||||||||||
Issuance under alternate compensation plan |
(f) | 18 | 15 | — | — | |||||||||||||||
Exercise of stock options |
1,730 | 1,113 | — | — | ||||||||||||||||
Stock-based compensation |
— | 869 | — | — | ||||||||||||||||
Balance at May 31, 2000 |
139,665 | 114,709 | 1,410 | 754 | ||||||||||||||||
Exercise of purchase warrants |
(d) | 168 | 93 | (168 | ) | (25 | ) | |||||||||||||
Issuance under alternate compensation plan |
(f) | 28 | 49 | — | — | |||||||||||||||
Exercise of stock options |
2,550 | 1,866 | — | — | ||||||||||||||||
Stock-based compensation |
— | 351 | — | — | ||||||||||||||||
Other |
— | 82 | — | — | ||||||||||||||||
Balance at May 31, 2001 |
142,411 | 117,150 | 1,242 | 729 | ||||||||||||||||
Exercise of compensation warrants |
(d) | 476 | 265 | (476 | ) | (70 | ) | |||||||||||||
Expiry of compensation warrants |
(e) | — | 659 | (766 | ) | (659 | ) | |||||||||||||
Exercise of stock options |
1,525 | 1,194 | — | — | ||||||||||||||||
Stock-based compensation |
— | (100 | ) | — | — | |||||||||||||||
Balance at May 31, 2002 |
144,412 | $ | 119,168 | 0 | $ | 0 | ||||||||||||||
| (b) 1997 Private Placement |
In May 2000, 892,857 common share purchase warrants related to an April 30, 1997 private placement were exercised to acquire 892,857 common shares at $1.68 per common share for aggregate cash proceeds of $1,500,000.
| (c) January 1999 Private Placement of Special Warrants |
On January 8, 1999, the Company completed a private placement of 5,333,333 special warrants for gross proceeds of $1,600,000 ($0.30 per special warrant) before deducting expenses of $383,000. Each special warrant granted the holder the right to acquire, without additional payment, one common share (stated capital $0.272 per common share) and one-half of one Series A purchase warrant (stated capital $0.028 per one-half common share purchase warrant). Each whole common share purchase warrant entitled the holder to acquire one common share for $0.36 at any time on or before January 8, 2000. On May 7, 1999 the special warrants were converted into 5,333,333 common shares and 2,666,667 purchase warrants. In addition, the Company granted 483,333 broker warrants and 50,000 compensation options (stated capital $0.12 per broker warrant and compensation option) to agents of the Company in connection with the completion of the offering. Each broker warrant and compensation option entitled the holder to acquire one common share for $0.30. All purchase warrants, broker warrants and compensation options related to this offering were exercised in the 2000 fiscal year.
74
| (d) October 1999 Private Placement of Special Warrants |
On October 27, 1999 the Company issued 30,303,031 special warrants for gross proceeds of $10,000,000 ($0.33 per special warrant) before deducting expenses of $1,562,000. The special warrants grant the holder the right to acquire, without additional payment, one common share of the Company (stated capital $0.316 per common share). The expenses include the issuance of 2,824,849 compensation warrants (stated capital $0.147 per warrant) for services in connection with the completion of the offering. Each compensation warrant entitles the holder to acquire one common share for $0.41 at any time prior to October 27, 2001. In the third quarter of 2000, the special warrants were converted into 30,303,031 common shares. During 2002, 475,700 compensation warrants were exercised. (2001 — 167,750 and 2000 — 2,181,399). As at May 31, 2002, no compensation warrants remain outstanding.
| (e) May 2000 Common Share Issue |
On May 2, 2000 the Company issued 15,333,334 common shares for gross proceeds of $46,000,000 ($3.00 per common share) before deducting expenses of $4,048,000. The expenses include the issuance of 766,666 compensation warrants (stated capital $0.86 per warrant) for services in connection with the completion of the offering. Each compensation warrant entitles the holder to acquire one common share for $3.30. The warrants vested 50% on November 2, 2000 and 50% on May 2, 2001. All compensation warrants expired unexercised on November 2, 2001.
| (f) Alternate Compensation Plans |
In 2000, the Company established a compensation plan for directors and officers, which allows the Company, in certain circumstances, to issue common shares to pay directors’ fees or performance bonuses of officers in lieu of cash. The number of common shares reserved for issuance under this plan is 2,500,000. Since inception, 46,000 shares have been issued under this plan.
The Company also established a deferred share unit plan that provides directors the option of receiving payment for their services in the form of share units rather than common shares or cash. Share units entitle the director to receive, on termination of their services to the Company, an equivalent number of common shares, or the cash equivalent of the market value of the common shares at that future date. The share units are granted based on the market value of the common shares on the date of issue. As of May 31, 2002 83,057 deferred share units have been issued, with a cash value of $62,000 being recorded in accrued liabilities.
75
| (g) Stock Option Plan |
Under the Company’s stock option plan, options may be granted to directors, officers, employees and consultants of the Company to purchase up to 12,000,000 common shares. Options are granted at the fair market value of the common shares on the date of grant. Options vest at various rates and have a term of five years. Stock option transactions for the three years ended May 31, 2002 are summarized as follows:
| 2002 | 2001 | 2000 | ||||||||||||||||||||||
| Weighted- | Weighted- | Weighted- | ||||||||||||||||||||||
| average | average | average | ||||||||||||||||||||||
| Options | exercise | Options | exercise | Options | exercise | |||||||||||||||||||
| (000's) | price | (000's) | price | (000's) | price | |||||||||||||||||||
Outstanding at beginning
of year |
4,144 | $ | 1.19 | 6,310 | $ | 0.80 | 3,094 | $ | 1.00 | |||||||||||||||
Granted |
3,188 | $ | 0.98 | 1,281 | $ | 2.08 | 5,135 | $ | 0.75 | |||||||||||||||
Exercised |
(1,525 | ) | $ | 0.78 | (2,550 | ) | $ | 0.73 | (1,730 | ) | $ | 0.64 | ||||||||||||
Forfeited |
(382 | ) | $ | 1.39 | (897 | ) | $ | 1.00 | (189 | ) | $ | 1.10 | ||||||||||||
Outstanding at end of year |
5,425 | $ | 1.17 | 4,144 | $ | 1.19 | 6,310 | $ | 0.80 | |||||||||||||||
Exercisable at end of year |
2,183 | $ | 1.32 | 2,486 | $ | 0.95 | 3,515 | $ | 0.78 | |||||||||||||||
The following table summarizes information about stock options outstanding at May 31, 2002:
| Options outstanding | Options exercisable | |||||||||||||||||||
| Weighted-average | ||||||||||||||||||||
| Options | remaining | Weighted- | Options | Weighted- | ||||||||||||||||
| outstanding | contractual life | average | exercisable | average | ||||||||||||||||
| Range of Exercise prices | (000's) | (years) | exercise price | (000's) | exercise price | |||||||||||||||
$0.33 to $0.49 |
609 | 2.3 | $ | 0.39 | 519 | $ | 0.39 | |||||||||||||
$0.50 to $0.99 |
3,368 | 4.0 | $ | 0.89 | 825 | $ | 0.86 | |||||||||||||
$1.00 to $1.99 |
625 | 3.4 | $ | 1.52 | 449 | $ | 1.50 | |||||||||||||
$2.00 to $3.63 |
823 | 3.3 | $ | 2.59 | 565 | $ | 2.58 | |||||||||||||
| 5,425 | $ | 1.17 | 2,358 | $ | 1.29 | |||||||||||||||
| (h) Deferred Stock-based Compensation |
The Company recorded deferred stock-based compensation recovery relating to options issued under the Company’s stock option plan amounting to $100,000 for the year ended May 31, 2002 (2001 — charge $351,000 and 2000 — charge $869,000). Amortization of deferred stock-based compensation was $296,000 for the year ended May 31, 2002 (2001 — $335,000 and 2000 — $330,000).
7. Income Taxes
Income tax recoveries attributable to losses from operations differ from the amounts computed by applying the combined Canadian federal and provincial income tax rates to pretax income from operations primarily as a result of the provision of a valuation allowance on net future income tax benefits.
76
Significant components of the Company’s future tax assets are as follows:
| As at May 31 (amounts in 000's) | 2002 | 2001 | ||||||
Non-capital loss carryforwards |
$ | 7,870 | $ | 9,976 | ||||
Research and development expenditures |
11,218 | 12,770 | ||||||
Book over tax depreciation |
1,537 | 1,819 | ||||||
Other |
787 | 1,984 | ||||||
Future tax assets |
21,412 | 26,549 | ||||||
Valuation allowance |
(21,412 | ) | (26,549 | ) | ||||
| $ | — | $ | — | |||||
In assessing the realizable benefit from future tax assets, management considers whether it is more likely than not that some portion or all of the future tax assets will not be realized. The ultimate realization of future tax assets is dependent on the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers projected future taxable income, uncertainties related to the industry in which the Company operates, and tax planning strategies in making this assessment. Due to the Company’s stage of development and operations, and uncertainties related to the industry in which the Company operates, the tax benefit of the above carried forward amounts have been completely offset by a valuation allowance.
Research and development expenditures can be carried forward indefinitely. To the extent that the non-capital loss carryforwards are not used, they expire as follows:
| Year of expiry (amounts in 000's) | Non-capital losses | |||
2003 |
$ | 2,140 | ||
2004 |
2,022 | |||
2005 |
2,295 | |||
2006 |
3,633 | |||
2007 |
3,630 | |||
2008 |
5,977 | |||
2009 |
5,627 | |||
| $ | 25,324 | |||
8. Research and Development Program
The Company’s cancer drug research and development program focuses primarily on the following technology platforms:
| (a) Immunotherapy |
This clinical approach stimulates the body’s natural defenses against cancer. The Company’s lead drug Virulizin® is currently in a North American Phase III clinical trial for the treatment of pancreatic cancer.
77
| (b) Antisense |
Antisense drugs are genetic molecules that inhibit the production of disease-causing proteins. GTI-2040 and GTI-2501, our lead antisense drugs, have shown pre-clinical anti-cancer activity across a broad range of cancers and are currently in phase II and phase I trials, respectively.
| (c) Small Molecules |
Anti-cancer activity was discovered with an anti-fungal agent Clotrimazole (“CLT”). Based on the structural feature found to be responsible for the anti-cancer effect of CLT, chemical analogues of CLT have been designed and tested. The lead analogue NC381 is in the pre-clinical stage of development.
| Period from | |||||||||||||||||||
| Year ended May 31 | inception | ||||||||||||||||||
| (amounts in 000's) | Sept. 5, 1986 to | ||||||||||||||||||
| Research and Development | 2002 | 2001 | 2000 | May 31, 2002 | |||||||||||||||
Immunotherapy |
|||||||||||||||||||
Acquired |
$ | — | $ | — | $ | — | $ | — | |||||||||||
Expensed |
4,612 | 2,161 | 887 | 29,488 | |||||||||||||||
Antisense |
|||||||||||||||||||
Acquired |
— | — | 11,000 | 11,000 | |||||||||||||||
Expensed |
3,410 | 7,116 | 2,772 | 13,298 | |||||||||||||||
Small Molecules
|
|||||||||||||||||||
Acquired |
— | — | — | 1,228 | |||||||||||||||
Expensed |
637 | 520 | 585 | 3,723 | |||||||||||||||
Total acquired |
$ | — | $ | — | $ | 11,000 | $ | 12,228 | |||||||||||
Total expensed |
$ | 8,659 | $ | 9,797 | $ | 4,244 | $ | 46,509 | |||||||||||
9. Supplementary Cash Flow Information
Changes in non-cash working capital balances for each of the periods ended are summarized as follows:
| Period from | ||||||||||||||||
| inception | ||||||||||||||||
| Sept. 5, 1986 to | ||||||||||||||||
| Years ended May 31 (amounts in 000's) | 2002 | 2001 | 2000 | May 31, 2002 | ||||||||||||
(Increase) decrease |
||||||||||||||||
Prepaid expenses and amounts receivable |
$ | 309 | $ | (409 | ) | $ | (440 | ) | $ | (618 | ) | |||||
Deferred charges |
— | — | 221 | — | ||||||||||||
Increase(decrease) |
||||||||||||||||
Accounts payable |
(2,686 | ) | 988 | 728 | (802 | ) | ||||||||||
Accrued liabilities |
253 | 1,269 | 66 | 2,750 | ||||||||||||
| $ | (2,124 | ) | $ | 1,848 | $ | 575 | $ | 1,330 | ||||||||
During the year ended May 31, 2002, the Company received interest of $2,488,000 (2001 — $2,607,000 and 2000 — $542,000).
78
10. Commitments
The Company has entered into operating leases for premises under which it is obligated to make minimum annual payments of approximately $210,000 in 2003, $230,000 in 2004 and $100,000 in 2005.
During the year ended May 31, 2002, operating lease expenses were $118,000 (2001 — $206,000 and 2000 — $146,000).
11. Related Party Transactions
During the year ended May 31, 2002, consulting fees of $68,000 were paid to individuals (or companies controlled by those individuals) who were either officers or directors of the Company (2001 — nil and 2000 — nil).
The Company received services from a law firm in which a director of the Company is a partner. Fees related primarily to consultations in the normal course of business for an aggregate of $376,000 for the year ended May 31, 2002 (2001 — $357,000 and 2000 — $425,000).
The amount payable to related parties as at May 31, 2002 was $46,000 (2001 — $140,000 and 2000 — $179,000).
12. Financial Instruments
The carrying values of cash and cash equivalents, short-term investments, accounts payable and accrued liabilities approximate their fair values due to the short-term nature of these instruments.
Financial instruments potentially exposing the Company to a concentration of credit risk consist principally of cash equivalents and short-term investments. The Company mitigates this risk by investing in high grade fixed income securities.
13. Canada and United States Accounting Policy Differences
These financial statements have been prepared in accordance with generally accepted accounting principles (“GAAP”) as applied in Canada. In certain respects, GAAP as applied in the United States differs from that applied in Canada.
| (a) SFAS 123 Employee Stock Compensation |
SFAS No. 123 encourages, but does not require, the recording of compensation costs for stock options issued to employees to be valued at fair value. For companies choosing not to adopt the fair value measurement for stock based compensation, the pronouncement requires the Company to disclose pro forma net income and earnings per share information as if the Company had accounted for its stock options under the fair value method since 1995. The Company has elected not to adopt the recording of compensation costs for stock options at fair value and, accordingly, a summary of the pro forma impact on the statement of loss is presented in the table below:
79
| (Amounts in 000's) | 2002 | 2001 | 2000 | |||||||||
Loss for the year |
$ | 13,487 | $ | 15,213 | $ | 8,599 | ||||||
Compensation expense related to the
fair value of stock options |
1,278 | 1,059 | 1,285 | |||||||||
Pro forma loss for the period |
$ | 14,765 | $ | 16,272 | $ | 9,884 | ||||||
Pro forma loss per common share |
$ | 0.10 | $ | 0.12 | $ | 0.11 | ||||||
The fair value of each option granted has been estimated at the date of grant using the Black-Scholes option pricing model with the following assumptions used for options granted in the years ended May 31, 2002, 2001, and 2000: (i) dividend yield of 0%; (ii) expected volatility of 80% (2001 — 95%, 2000 — 95%); (iii) risk-free interest rate of 3.6% (2001 — 5.4%, 2000 — 6.0%) and (iv) expected lives of 5 years. The Company has assumed no forfeiture rate as adjustments for actual forfeitures are made in the year they occur. The weighted-average grant-date fair values of options issued in the years ended May 31, 2002, 2001, and 2000 were $ 0.71, $1.56, and $0.60 respectively.
| (b) SFAS 130 Reporting Comprehensive Income |
SFAS No. 130 establishes standards for reporting and presentation of comprehensive income. This standard defines comprehensive income as the changes in equity of an enterprise except those resulting from shareholder transactions. Comprehensive loss for the periods presented in these financial statements equaled the loss for the period.
Item 18. Financial Statements
The Company has elected to report under Item No. 17.
80
GLOSSARY
The following is a glossary of terms that are used in this Annual Report
| Actinic keratosis: | a condition that arises on the skin’s surface. It can be the first step in the development of skin cancer and therefore is a precursor of cancer, or a precancer. | |
| Analogue: | a chemical derivative or variation of a parent molecule | |
| Anti-angiogenic: | preventing blood vessel formation | |
| Anti-metastatic | the ability to inhibit the movement of tumor cells from a primary/original site to other organs in the body | |
| Anti-proliferative: | preventing cancer cell division | |
| Apoptosis: | programmed cell death | |
| BCD: | Bureau of Control of Drugs, the regulatory agency controlling pharmaceutical drugs in Mexico | |
| Biologic response modifier or BRM: |
a substance which stimulates, modifies or enhances the body’s response, including the response of the body’s immune and other protective cellular and molecular systems, to certain diseases | |
| Carcinoma: | any cancerous tumor that starts with the cells that cover the inner and outer body surfaces | |
| Clinical trials: | the investigational use of a new drug in humans: Phase I clinical trials test a drug for safety, Phase II clinical trials test a drug for efficacy and safety in a relatively small sample of patients, and Phase III clinical trials test the drug for efficacy in larger numbers of patients and compares the drug with conventional therapies | |
| cGMP: | current good manufacturing practices, as mandated from time to time by the TPP and the FDA | |
| CLT: | Clotrimazole | |
| Cytokine: | a generic term for a non-antibody protein released by a cell population (e.g., activated macrophages) of the immune system on contact with chemical or biological stimuli | |
| Cytostatic: | pertaining to the inhibition of cell growth without cell destruction | |
| Cytotoxic: | pertaining to the destruction of cells | |
| Deoxyribonucleic acid (DNA): |
DNA is the carrier of genetic information which exists in all cells of the body. The building blocks of DNA are called nucleotides | |
| Efficacy: | the ability of a drug to produce a desired result | |
| EMEA: | European Medicine Evaluation Agency | |
| FDA: | Food and Drug Administration, the government agency which regulates the use and sale of diagnostic and therapeutic drug products in the United States | |
| Gene expression: | the synthesis of specific proteins on the basis of inherited or acquired genetic information | |
| GeneSense: | GeneSense Technologies Inc., a subsidiary of the Company | |
| HIV: | Human Immunodeficiency Virus, the virus which causes AIDS |
81
| Immune system: | the totality of organs and cells involved in the body’s immunologic response to foreign antigens and malignant tissue | |
| IND: | investigational new drug | |
| In situ: | in position | |
| In vitro: | in the test tube; referring to chemical reactions, fermentation, etc., occurring therein e.g., in cell-free extracts | |
| In vivo: | in the living body; referring to chemical processes occurring within cells, etc., as distinguished from those occurring in cell-free extracts (in vitro) | |
| LD50: | the measure (quantity) of a drug that, when administered to experimental animals in acute toxicity studies, is lethal to 50 percent of such animals | |
| Macrophage: | a large scavenger white blood cell that engulfs and digests invading micro-organisms and cell debris, and also participates in many complex immunologic processes | |
| Malignant/ malignancy: |
describes a tumor that is cancerous. Two important qualities of malignancies are the tendency to invade surrounding tissues and to break off and spread elsewhere (metastasis) | |
| MAP Kinase Pathway: | the pathway of mitogenic signal transduction through the cascade of mitogen-activated protein (MAP) kinases which ultimately lead to alteration in regulatory events such as cell proliferation, differentiation and apoptosis. | |
| Metabolism: | the overall biochemical reactions that take place in a living organism including the building up of complex molecules or breakdown of molecules to provide energy | |
| Metastasis: | the process by which tumor cells are spread to other parts of the body | |
| mRNA: | messenger, or mRNA, is a copy of the information carried by a gene on the DNA. The role of mRNA is to move the information contained in DNA to the translation machinery. | |
| NDA: | new drug application, the application to obtain marketing approval filed with the FDA or BCD after completion of human clinical trials | |
| NDS: | new drug submission, the application to obtain marketing approval filed with the TPP after completion of human clinical trials | |
| NuChem: | NuChem Pharmaceuticals Inc., a subsidiary of the Company | |
| NuChem Analogues: | analogues of CLT licensed by the Company for anti-cancer indications | |
| Nucleic acid: | DNA and RNA, each of which are formed by the combination of nucleotides; it is found in all living cells and contains the genetic code required to transfer genetic information from one generation to the next | |
| Nucleotide: | a compound consisting of a purine or pyrimidine base, a pentose sugar and a phosphoric acid; they are the building blocks from which nucleic acids (DNA or RNA) are constructed | |
| Oligonucleotides: | oligonucleotides are short chains of nucleotides, which are the building blocks of DNA and RNA | |
| Peptide: | a molecule containing two or more amino acids, most frequently linked in a single chain; amino acids form the constituent parts of proteins (see protein) |
82
| Pharmacokinetics: | the action of drugs in the body over a period of time, including the process of absorption, distribution, localization in tissues, biotransformation and excretion | |
| Pre-clinical testing: | testing that is conducted in the laboratory (chemistry and pharmacology) and with animals to help determine a product’s chemical, pharmacological and pharmaceutical characteristics (including mechanism of action), toxicity, efficacy and side effects | |
| Proteins: | large molecules composed of long chains of sub-units of amino acids | |
| R1 and R2: | components of ribonucleotide reductase | |
| Ribonucleic acid (RNA): |
a nucleic acid found in both the nucleus and the cytoplasm of all cells. It carries genetic information from the nucleus to the cytoplasm, where it also reacts as a template in association with ribosomes to synthesize proteins | |
| Salvage therapy: | therapies for patients who have exhausted all standard treatment options | |
| SCID: | severe combined immunodeficiency disease | |
| Squamous cell carcinoma: |
second most common skin cancer | |
| SSA: | Secretaria de Salud (the Ministry of Health for Mexico) | |
| Stage IV cancer: | distant metastatic cancer spread | |
| Toxicity: | a condition that results from exposure to a substance at levels causing deleterious side effects which may be harmful to an organism | |
| TPP: | Therapeutic Products Programme. A division of the Health Protection Branch, Department of National Health and Welfare (Canada), the government agency which regulates the use and sale of therapeutic drug products in Canada | |
| Tumor: | an abnormal swelling or lump in the body caused by the growth of new tissues which differ in structure from the part of the body in which they are growing. A tumor may be benign or malignant | |
| Tumor necrosis: | tumor deterioration and death | |
| Xenograft: | an implant of a foreign substance |
VIRULIZIN® is a trademark of the Company. All other trademarks or trade names referred to in this Annual Report are the property of their respective owners.
83
Item 19. EXHIBITS
Report of Independent Chartered Accountants.
The following exhibits are filed as part of this Annual Report.
| * | 1.1 | Articles of Amalgamation. | ||
| * | 1.2 | Bylaws. |
||
| ** | 1.3 | Articles of Amendment. |
||
| **** | 1.4 | Articles of Amendment, dated November 27, 1996, regarding name change of Company to Imutec Pharma Inc. |
||
| ***** | 1.5 | Articles of Amendment dated November 19, 1998, regarding name change of Company to Lorus Therapeutics Inc. |
||
| * | 2.1 | Form of stock option agreement for officers, directors and employees. |
||
| *** | 2.2 | Stock Option Plan of the Company dated June 3, 1993, as amended. |
||
| **** | 2.3 | Stock Option Plan of the Company dated December 11, 1997, as amended. |
||
| ***** | 2.4 | Stock Option Plan of the Company dated November 19, 1998, as amended. |
||
| * | 4.1 | Amalgamation Agreement dated August 23, 1991, among the Company, Mint Gold Resources Ltd., Harry J. Hodge and Wayne Beach. | ||
| 4.2 | Lease of Premises between the Company and 565991 Ontario Limited dated July 27, 2001. |
|||
| * | 4.3 | Shareholders Agreement dated October 2, 1989. |
||
| * | 4.4 | Shareholders Agreement termination agreement dated July 29, 1991. |
||
| ** | 4.5 | Amended and Restated Escrow Agreement dated as of October 28, 1991 among the Company, Montreal Trust Company of Canada and certain shareholders of the Company. | ||
| ** | 4.6 | Offer to Lease dated May 11, 1992 between the Company and Morningside Properties Limited, as amended on May 26, 1992. | ||
| 10.1 | Certification pursuant to 18 USC. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|||
| 10.2 | Certification pursuant to 18 USC. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| * | Incorporated by reference to File 0-19763, Registration Statement on Form 20-FR, dated March 4, 1992. | |
| ** | Incorporated by reference to File 0-19763, Annual Report on Form 20-F, dated September 28, 1992. | |
| *** | Incorporated by reference to File 0-19763, Annual Report on Form 20-F, dated December 14, 1995. | |
| **** | Incorporated by reference to File 0-19763, Annual Report on Form 20-F, dated November 18, 1998. | |
| ***** | Incorporated by reference to File 0-1963, Annual Report on Form 20-F dated November 30, 1999. |
84
SIGNATURES
The Company hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this registration statement [annual report] on its behalf.
| Lorus Therapeutics Inc. | ||
|
|
||
| (Company) | ||
| /s/ Jim A.
Wright Jim A. Wright |
||
| Chief Executive Officer | ||
| Date: December 13, 2002 |
CERTIFICATIONS
I, Jim A. Wright certify that:
| 1. | I have reviewed this annual report on Form 20-F of Lorus Therapeutics Inc.; | ||
| 2. | Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report; and | ||
| 3. | Based on my knowledge, the financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this annual report; |
| Date: December 13, 2002 | ||
| /s/ Jim A. Wright Jim A. Wright |
||
| Chief Executive Officer |
85
CERTIFICATIONS
I, James T. Parsons certify that:
| 1. | I have reviewed this annual report on Form 20-F of Lorus Therapeutics Inc.; | ||
| 2. | Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report; and | ||
| 3. | Based on my knowledge, the financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this annual report; |
| Date: | December 13, 2002 | |||
| /s/ James T. Parsons James T. Parsons |
||||
| VP Finance and Administration and Chief Financial Officer |
86